
Whole Exome Sequencing Reveals Clustering of Variants of Known Vitiligo Genes in Multiplex Consanguineous Pakistani Families

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Family Enrollment and Clinical Classification
2.2. Sample Collection and Isolation of DNA
2.3. Whole Exome Sequencing
2.4. Molecular Modeling
3. Results
3.1. Clinical Evaluation of Vitiligo in Multiplex Pakistani Families
3.2. Genetic Analysis of Families with Vitiligo
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berqvist, C.; Ezzedine, K. Vitiligo: A Review. Dermatology 2020, 236, 571–592. [Google Scholar] [CrossRef]
- Garcia-Melendez, M.E.; Salinas-Santander, M.; Sanchez-Dominguez, C.; Gonzalez-Cardenas, H.; Cerda-Flores, R.M.; Ocampo-Candiani, J.; Ortiz-López, R. Protein tyrosine phosphatase PTPN22 +1858C/T polymorphism is associated with active vitiligo. Exp. Med. 2014, 8, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Spritz, R.A. Recent progress in the genetics of generalized vitiligo. J. Genet. Genom. 2011, 38, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Czajkowski, R.; Mecinska-Jundzill, K. Current aspects of vitiligo genetics. Postep. Derm. Alergol. 2014, 31, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.H.L.; Paul, S.; Yorgov, D.; Santorico, S.A.; Spritz, R.A. Family Clustering of Autoimmune Vitiligo Results Principally from Polygenic Inheritance of Common Risk Alleles. Am. J. Hum. Genet. 2019, 105, 364–372. [Google Scholar] [CrossRef]
- Kim, S.M.; Chung, H.S.; Hann, S.-K. The genetics of vitiligo in Korean patients. Int. J. Dermatol. 1998, 37, 908–910. [Google Scholar] [CrossRef]
- Laddha, N.C.; Dwivedi, M.; Mansuri, M.S.; Gani, A.R.; Ansarullah, M.; Ramachandran, A.V.; Dalai, S.; Begum, R. Vitiligo: Interplay between oxidative stress and immune system. Exp. Derm. 2013, 22, 245–250. [Google Scholar] [CrossRef]
- Yu, S.; Doycheva, D.M.; Gamdzyk, M.; Yang, Y.; Lenahan, C.; Li, G.; Li, D.; Lian, L.; Tang, J.; Lu, J.; et al. Activation of MC1R with BMS-470539 attenuates neuroinflammation via cAMP/PKA/Nurr1 pathway after neonatal hypoxic-ischemic brain injury in rats. J. Neuroinflamm. 2021, 18, 26. [Google Scholar] [CrossRef]
- Alikhan, A.; Felsten, L.M.; Daly, M.; Petronic-Rosic, V. Vitiligo: A comprehensive overview Part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J. Am. Acad. Derm. 2011, 65, 473–491. [Google Scholar] [CrossRef]
- Noman, M.; Ishaq, R.; Bukhari, S.A.; Ahmed, Z.M.; Riazuddin, S. Delineation of Homozygous Variants Associated with Prelingual Sensorineural Hearing Loss in Pakistani Families. Genes 2019, 10, 1031. [Google Scholar] [CrossRef]
- Richard, E.M.; Santos-Cortez, R.L.P.; Faridi, R.; Rehman, A.U.; Lee, K.; Shahzad, M.; Acharya, A.; Khan, A.A.; Imtiaz, A.; Chakchouk, I.; et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum. Mutat. 2019, 40, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Riazuddin, S.; Hussain, M.; Razzaq, A.; Iqbal, Z.; Shahzad, M.; Polla, D.L.; Song, Y.; van Beusekom, E.; Khan, A.A.; Tomas-Roca, L.; et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol. Psychiatry 2017, 22, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Jaworek, T.J.; Kausar, T.; Bell, S.M.; Tariq, N.; Maqsood, M.I.; Sohail, A.; Ali, M.; Iqbal, F.; Rasool, S.; Riazuddin, S.; et al. Molecular genetic studies and delineation of the oculocutaneous albinism phenotype in the Pakistani population. Orphanet. J. Rare Dis. 2012, 7, 44. [Google Scholar]
- Debasis, d.; Vivek; Manchanda; Singh, P.; Ankita; Gupta, I. A Compendium of Genes and Variations Associated with Vitiligo. 2023. Available online: https://vitivar.igib.res.in/genes.php (accessed on 12 March 2023).
- Canton, I.; Akhtar, S.; Gavalas, N.G.; Gawkrodger, D.J.; Blomhoff, A.; Watson, P.F.; Weetman, A.P.; Kemp, E.H. A single-nucleotide polymorphism in the gene encoding lymphoid protein tyrosine phosphatase (PTPN22) confers susceptibility to generalised vitiligo. Genes Immun. 2005, 6, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Passeron, T.; Ortonne, J.P. Activation of the unfolded protein response in vitiligo: The missing link? J. Investig. Derm. 2012, 132, 2502–2504. [Google Scholar] [CrossRef]
- Tang, X.; Fang, F.; Yang, J.; Zheng, X.; Fan, M.; Wang, L.; Zhang, A. Association Study Reveals One Susceptibility Locus with Vitiligo in the Chinese Han Population. Genet. Test. Mol. Biomark. 2019, 23, 791–796. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Z.; Chai, L.; Che, Y.; Min, S.; Yang, R. Identification and characterization of a unique leucine-rich repeat protein (LRRC33) that inhibits Toll-like receptor-mediated NF-κB activation. Biochem. Biophys. Res. Commun. 2013, 434, 28–34. [Google Scholar] [CrossRef]
- Rajendiran, K.S.; Rajappa, M.; Chandrashekar, L.; Thappa, D.M. Association of PTPN22 gene polymorphism with non-segmental vitiligo in South Indian Tamils. Postep. Derm. Alergol. 2018, 35, 280–285. [Google Scholar] [CrossRef]
- Cheong, K.A.; Kim, N.H.; Noh, M.; Lee, A.Y. Three new single nucleotide polymorphisms identified by a genome-wide association study in Korean patients with vitiligo. J. Korean Med. Sci. 2013, 28, 775–779. [Google Scholar] [CrossRef]
- Rodrigues, M.; Ezzedine, K.; Hamzavi, I.; Pandya, A.G.; Harris, J.E.; Vitiligo Working Group. New discoveries in the pathogenesis and classification of vitiligo. J. Am. Acad. Dermatol. 2017, 77, 1–13. [Google Scholar] [CrossRef]
- Laddha, N.C.; Dwivedi, M.; Shajil, E.M.; Prajapati, H.; Marfatia, Y.S.; Begum, R. Association of PTPN22 1858C/T polymorphism with vitiligo susceptibility in Gujarat population. J. Derm. Sci. 2008, 49, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Rendtlew Danielsen, J.; Fugger, K.; Gromova, I.; Nerstedt, A.; Lukas, C.; Bartek, J.; Lukas, J.; Mailand, N. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell. Biol. 2010, 12, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Ferrara, T.M.; Ben, S.; Riccardi, S.L.; Cole, J.B.; Gowan, K.; Holland, P.J.; Bennett, D.C.; et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 2012, 44, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Andersen, G.H.L.; Santorico, S.A.; Spritz, R.A. Multiple Functional Variants of IFIH1, a Gene Involved in Triggering Innate Immune Responses, Protect against Vitiligo. J. Invest. Derm. 2017, 137, 522–524. [Google Scholar] [CrossRef]
- Kundu, R.V.; Mhlaba, J.M.; Rangel, S.M.; Le Poole, I.C. The convergence theory for vitiligo: A reappraisal. Exp. Dermatol. 2019, 28, 647–655. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
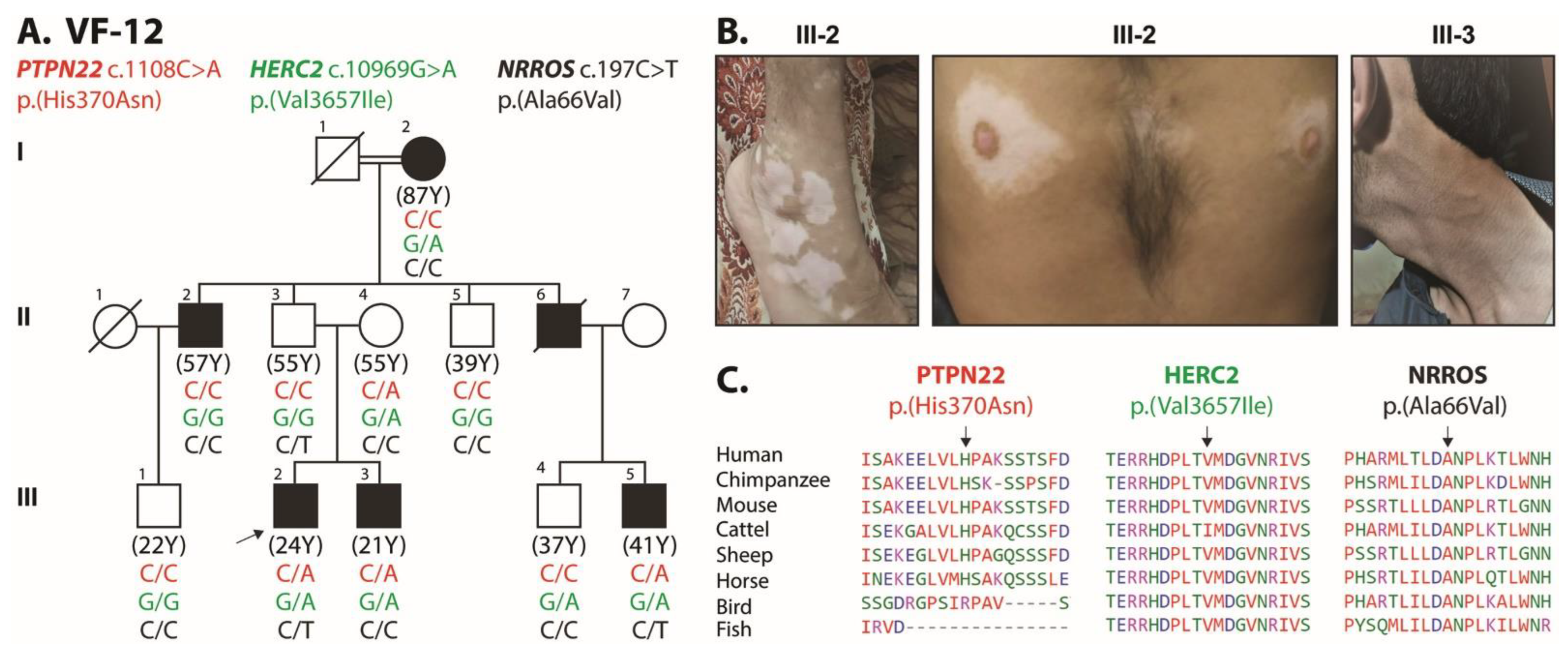{kind=link}
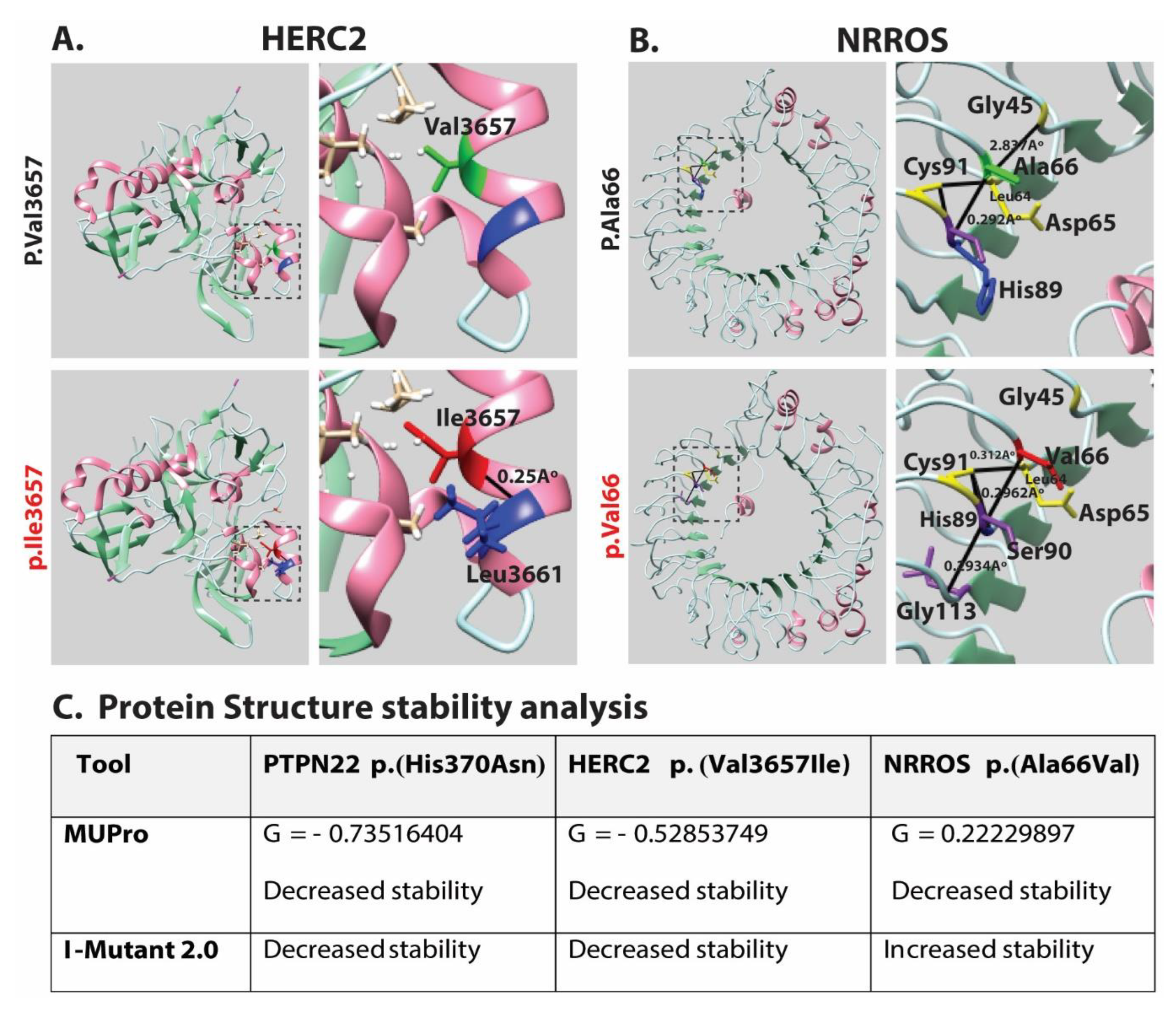{kind=link}
{kind=link}
| Family ID | Family Ethnicity | Individual ID | Age (Year) | Age of Onset | Gender | Phenotype | Doctoral Description of the Disease | Affected Area of Skin (%) | Comorbidity |
|---|---|---|---|---|---|---|---|---|---|
| VF-01 | Rajput | VF01_01 | 28 | 18 | Male | Affected | NSV, Generalized vitiligo | 20–30% | Nil |
| VF01_02 | 26 | 21 | Female | Affected | NSV, Generalized Vitiligo | 20–30% | Nil | ||
| VF01_04 | 17 | 14 | Female | Affected | NSV, Generalized vitiligo | 10–20% | Nil | ||
| VF-02 | Mughal | VF02_03 | 28 | 20 | Female | Affected | NSV, Generalized vitiligo | 10–20% | Nil |
| VF02_06 | 18 | 18 | Female | Affected | NSV, Generalized Vitiligo | Less than 5% | Nil | ||
| VF02_07 | 42 | 39 | Male | Recovered | Generalized Vitiligo (NSV*) | N/A | Nil | ||
| VF02_11 | 32 | 17 | Male | Affected | NSV, Generalized vitiligo | 30–40% | Nil | ||
| VF02_15 | 80 | 14 | Female | Affected | Vitiligo Universalis | 90–100% | Diabetes | ||
| VF02_16 | 79 | 19 | Male | Affected | NSV, Generalized vitiligo | 90–100% | Nil | ||
| VF02_17 | 56 | 18 | Female | Affected | NSV, Generalized vitiligo | 90–100% | Nil | ||
| VF02_18 | 23 | 11 | Female | Affected | NSV, Generalized vitiligo | 10–20% | Nil | ||
| VF02_19 | 20 | 10 | Female | Recovered | Vitiligo recovered (NSV*) | N/A | Nil | ||
| VF-03 | Awan | VF03_01 | 46 | 24 | Male | Affected | NSV, Generalized vitiligo | 30–40% | Nil |
| VF03_07 | 7 | 04 | Male | Affected | NSV, Acrofacial vitiligo | 5–10% | Nil | ||
| VF03_08 | 30 | 27 | Female | Affected | NSV, Acrofacial vitiligo | Less than 5% | Nil | ||
| VF-04 | Sardar | VF04_01 | 30 | 24 | Male | Affected | NSV, Generalized vitiligo | 20–30% | Nil |
| VF04_02 | 38 | 31 | Male | Affected | NSV, Generalized vitiligo | Less than 5% | Nil | ||
| VF04_03 | 13 | 7 | Female | Affected | NSV, Generalized vitiligo | Less than 5% | Nil | ||
| VF-05 | Mughal | VF05_01 | 47 | 33 | Male | Affected | NSV, Generalized vitiligo | 20–30% | Nil |
| VF05_04 | 22 | 21 | Male | Affected | Vitiligo started in neck region | Less than 5% | Nil | ||
| VF-06 | Rajput | VF06_01 | 31 | 11 | Male | Affected | NSV, Generalized vitiligo | 70–80% | Nil |
| VF06_02 | 25 | 13 | Male | Affected | NSV, Generalized vitiligo | 40–50% | Nil | ||
| VF-07 | Niazi | VF07_01 | 55 | 23 | Male | Affected | NSV, Generalized vitiligo | 40–50% | Nil |
| VF07_03 | 21 | 19 | Male | Affected | NSV, Generalized vitiligo | 5–10% | Nil | ||
| VF-08 | Shiekh | VF08_01 | 69 | 19 | Female | Affected | NSV, Generalized vitiligo | 20–30% | Nil |
| VF08_04 | 21 | 17 | Male | Affected | NSV, Generalized vitiligo | 5–10% | Nil | ||
| VF-09 | Awan | VF09_01 | 64 | 59 | Female | Affected | NSV, Generalized vitiligo | 20–30% | Nil |
| VF09_05 | 17 | 15 | Male | Affected | NSV, Generalized vitiligo | Less than 5% | Nil | ||
| VF09_07 | 19 | 15 | Female | Affected | NSV, Generalized vitiligo | Less than 5% | Nil | ||
| VF09_10 | 9 | 2 | Female | Affected | NSV, Generalized vitiligo | 20–30% | Nil | ||
| VF-10 | Malik | VF10_01 | 79 | 33 | Male | Affected | NSV, Generalized vitiligo | 60–70% | Nil |
| VF10_04 | 83 | 42 | Male | Affected | NSV, Generalized vitiligo | 70–80% | Nil | ||
| VF10_07 | 80 | 40 | Female | Affected | NSV, Generalized vitiligo | 40–50% | Nil | ||
| VF10_10 | 35 | 27 | Male | Affected | NSV, Generalized vitiligo | 40–50% | Nil | ||
| VF10_11 | 51 | 33 | Male | Affected | NSV, Generalized vitiligo | 50–60% | Nil | ||
| VF10_14 | 12 | 7 | Male | Affected | NSV, Generalized vitiligo | 5–10% | Nil | ||
| VF10_16 | 28 | 17 | Male | Affected | NSV, Generalized vitiligo | 40–50% | Nil | ||
| VF-11 | Qazi | VF11_01 | 56 | 11 | Female | Affected | Vitiligo Universalis | 90–100% | Nil |
| VF11_02 | 58 | 53 | Male | Affected | Vitiligo just started at the skull | Less than 1% | Nil | ||
| VF11_03 | 19 | 11 | Male | Affected | NSV, Generalized vitiligo | 5–10% | Nil | ||
| VF-12 | Malik | VF12_01 | 57 | 43 | Male | Affected | NSV, Generalized vitiligo | 30–40% | Diabetes |
| VF12_05 | 26 | 15 | Male | Affected | NSV, Generalized vitiligo | 30–40% | Nil | ||
| VF12_06 | 23 | 22 | Male | Affected | NSV, Generalized vitiligo | Less than 5% | Nil | ||
| VF12_07 | 35 | 24 | Male | Affected | NSV, Generalized vitiligo | 20–30% | Nil | ||
| VF12_09 | 79 | 68 | Female | Affected | NSV, Generalized vitiligo | 5–10% | Nil | ||
| VF-13 | Rajput | VF13_01 | 74 | 66 | Male | Affected | NSV, Generalized vitiligo | 30–40% | Nil |
| VF13_04 | 13 | 9 | Male | Affected | NSV, Generalized vitiligo | 5–10% | Nil | ||
| VF-14 | Chaudary | VF14_01 | 67 | 31 | Female | Affected | NSV, Generalized vitiligo | 15–20% | Nil |
| VF14_03 | 47 | 38 | Male | Affected | NSV, Generalized vitiligo | 30–40% | Nil | ||
| VF14_04 | 15 | 9 | Female | Affected | NSV, Generalized vitiligo | 10–20% | Nil | ||
| VF-15 | Rajput | VF15_01 | 77 | 19 | Male | Affected | Vitiligo Universalis | 90–100% | Nil |
| VF15_04 | 21 | 14 | Male | Affected | NSV, Generalized vitiligo | 20–30% | Nil | ||
| VF15_07 | 21 | 13 | Male | Affected | NSV, Generalized vitiligo | 30–40% | Nil |
| Family | Chr | Gene | Ref | Alt | c.DNA Change | Protein Change | SNP ID | rs Number | ExAC | CADD | SIFT | Polyphen | Mutation Taster | Mutation Assessor | Genotyping of Proband |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VF-01 | 2 | IFIH1 | T | C | c.A1046G | p.K349R | NM_022168 | rs72650664 | 0.0035 | 14.59 | T | B | D | N | Het |
| 11 | GSTP1 | A | C | c.A601C | p.N201H | NM_000852 | rs145957405 | - | 12.59 | D | P | D | M | Het | |
| 11 | GSTP1 | A | C | c.A602C | p.N201T | NM_000852 | rs376074280 | 0.00001677 | 7.554 | T | B | D | M | Het | |
| 12 | ATXN2 | TGC | - | c.534_536del | p.178_179del | NM_002973 | rs193922927 | 0 | - | - | - | - | - | Het | |
| 15 | HERC2 | CT | - | c.5780_5781de | p.K1927fs | NM_004667 | - | - | - | - | - | - | - | Het | |
| 16 | RPGRIP1L | T | A | c.A278T | p.E93V | NM_015272 | rs765876839 | 0.00000825 | 18.56 | T | P | D | L | Het | |
| X | IL1RAPL1 | TG | - | c.63_64del | p.V21fs | NM_014271 | rs147274241 | . | . | . | . | . | . | Het | |
| VF-02 | 19 | BCL2L12 | G | A | c.G542A | p.R181Q | NM_138639 | rs762286281 | 0.0003 | 19.56 | T | D | D | L | Het |
| VF-03 | 8 | TG | C | T | c.C3902T | p.P1301L | NM_003235 | rs549184203 | 0.0002 | 21.1 | D | D | D | M | Het |
| 11 | GSTP1 | A | G | c.A1G | p.M1V | NM_000852 | rs1448870282 | - | 10.05 | D | B | - | - | Het | |
| VF-09 | 3 | GPX1 | C | A | c.G442T | p.A148S | NM_001329455 | rs6446261 | - | 6.871 | T | B | D | L | Het |
| 3 | GPX1 | GCC | - | c.33_35del | p.11_12del | NM_001329455 | rs17838762 | - | - | - | - | - | - | Het | |
| 9 | TLR4 | A | G | c.A1084G | p.K362E | NM_003266 | rs539153708 | 0.0002 | 12.84 | T | P | N | N | Het | |
| 10 | CASP7 | C | T | c.C481T | p.R161W | NM_033340 | rs185649982 | 0 | 0 | T | B | N | - | Het | |
| 21 | UBASH3A | G | A | c.G1666A | p.E556K | NM_018961 | rs116752327 | 0.0001 | 15.6 | D | D | D | M | Het | |
| VF-10 | 1 | PTPRC | T | A | c.T566A | p.I189N | NM_002838 | rs201715157 | 0.0013 | 7.444 | D | B | N | L | Het |
| 2 | FARP2 | A | G | c.A134G | p.H45R | NM_014808 | rs150312458 | 0.0034 | 8.045 | T | B | N | N | Het | |
| 8 | TG | G | A | c.G2963A | p.R988H | NM_003235 | rs16893332 | 0.0062 | 11.02 | T | B | P | N | Het | |
| 14 | STRN3 | G | T | c.C53A | p.P18H | NM_001083893 | rs1036923688 | . | 15.33 | D | B | D | N | Het | |
| 16 | CDH1 | G | A | c.G188A | p.R63Q | NM_004360 | rs587780117 | 0.00005765 | 12.03 | T | B | N | M | Het | |
| 17 | NOS2 | G | A | c.C2084T | p.P695L | NM_000625 | rs767807366 | 0.00004954 | 19.85 | T | P | D | M | Het | |
| VF-12 | 1 | PTPN22 | G | T | c.C1108A | p.H370N | NM_015967 | rs72650671 | 0.0023 | 6.752 | T | P | N | M | Het |
| 10 | ACTA2 | C | T | - | Splicing | - | rs112687898 | - | 26 | - | - | D | - | Het | |
| 15 | HERC2 | C | T | c.G10969A | p.V3657I | NM_004667 | rs139953376 | 0.0025 | 3.685 | T | B | N | N | Het | |
| 20 | RALY | G | A | c.G694A | p.G232S | NM_016732 | rs779745009 | 0.0085 | 7.476 | T | B | N | N | Het | |
| VF-15 | 1 | PTPRC | A | G | c.A1808G | p.D603G | NM_002838 | rs754699279 | 0.00004967 | 12.77 | T | P | D | L | Het |
| 3 | TGFBR2 | G | A | c.G1283A | p.R428H | NM_001024847 | rs143095746 | 0.0004 | 17.14 | D | B | D | L | Het | |
| 3 | GPX1 | GCC | - | c.33_35del | p.11_12del | NM_001329455 | rs17838762 | - | - | - | - | - | - | Het |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishaq, R.; Ilyas, M.; Habiba, U.; Amin, M.N.u.; Saeed, S.; Raja, G.K.; Shaiq, P.A.; Ahmed, Z.M. Whole Exome Sequencing Reveals Clustering of Variants of Known Vitiligo Genes in Multiplex Consanguineous Pakistani Families. Genes 2023, 14, 1118. https://doi.org/10.3390/genes14051118
Ishaq R, Ilyas M, Habiba U, Amin MNu, Saeed S, Raja GK, Shaiq PA, Ahmed ZM. Whole Exome Sequencing Reveals Clustering of Variants of Known Vitiligo Genes in Multiplex Consanguineous Pakistani Families. Genes. 2023; 14(5):1118. https://doi.org/10.3390/genes14051118
Chicago/Turabian StyleIshaq, Rafaqat, Muhammad Ilyas, Umme Habiba, Muhammad Noor ul Amin, Sadia Saeed, Ghazala Kaukab Raja, Pakeeza Arzoo Shaiq, and Zubair M. Ahmed. 2023. "Whole Exome Sequencing Reveals Clustering of Variants of Known Vitiligo Genes in Multiplex Consanguineous Pakistani Families" Genes 14, no. 5: 1118. https://doi.org/10.3390/genes14051118
APA StyleIshaq, R., Ilyas, M., Habiba, U., Amin, M. N. u., Saeed, S., Raja, G. K., Shaiq, P. A., & Ahmed, Z. M. (2023). Whole Exome Sequencing Reveals Clustering of Variants of Known Vitiligo Genes in Multiplex Consanguineous Pakistani Families. Genes, 14(5), 1118. https://doi.org/10.3390/genes14051118