
Fecal Microbial Structure and Metabolic Profile in Post-Weaning Diarrheic Piglets
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Study Design and Animals
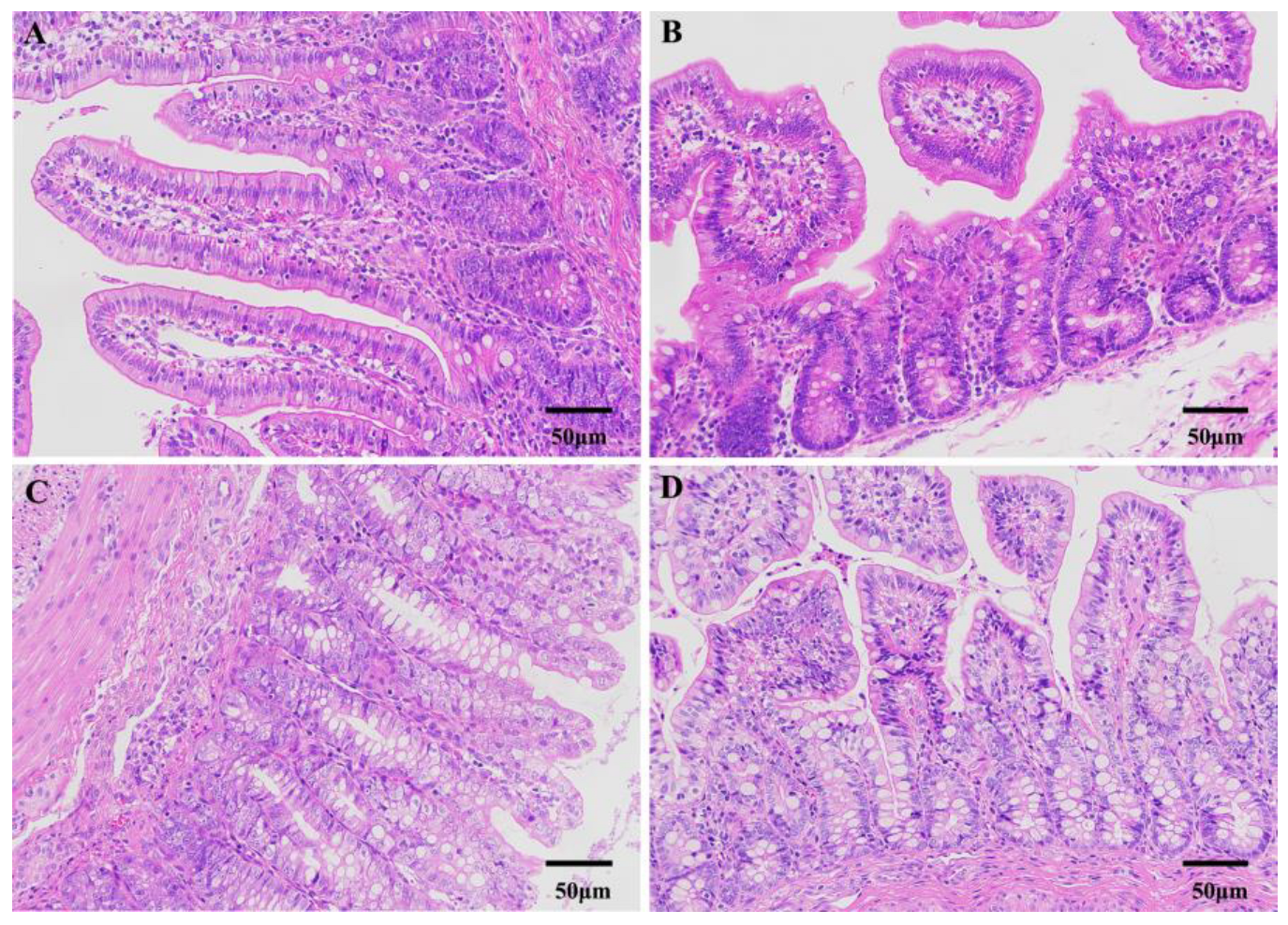
2.3. Intestinal Morphology Detection
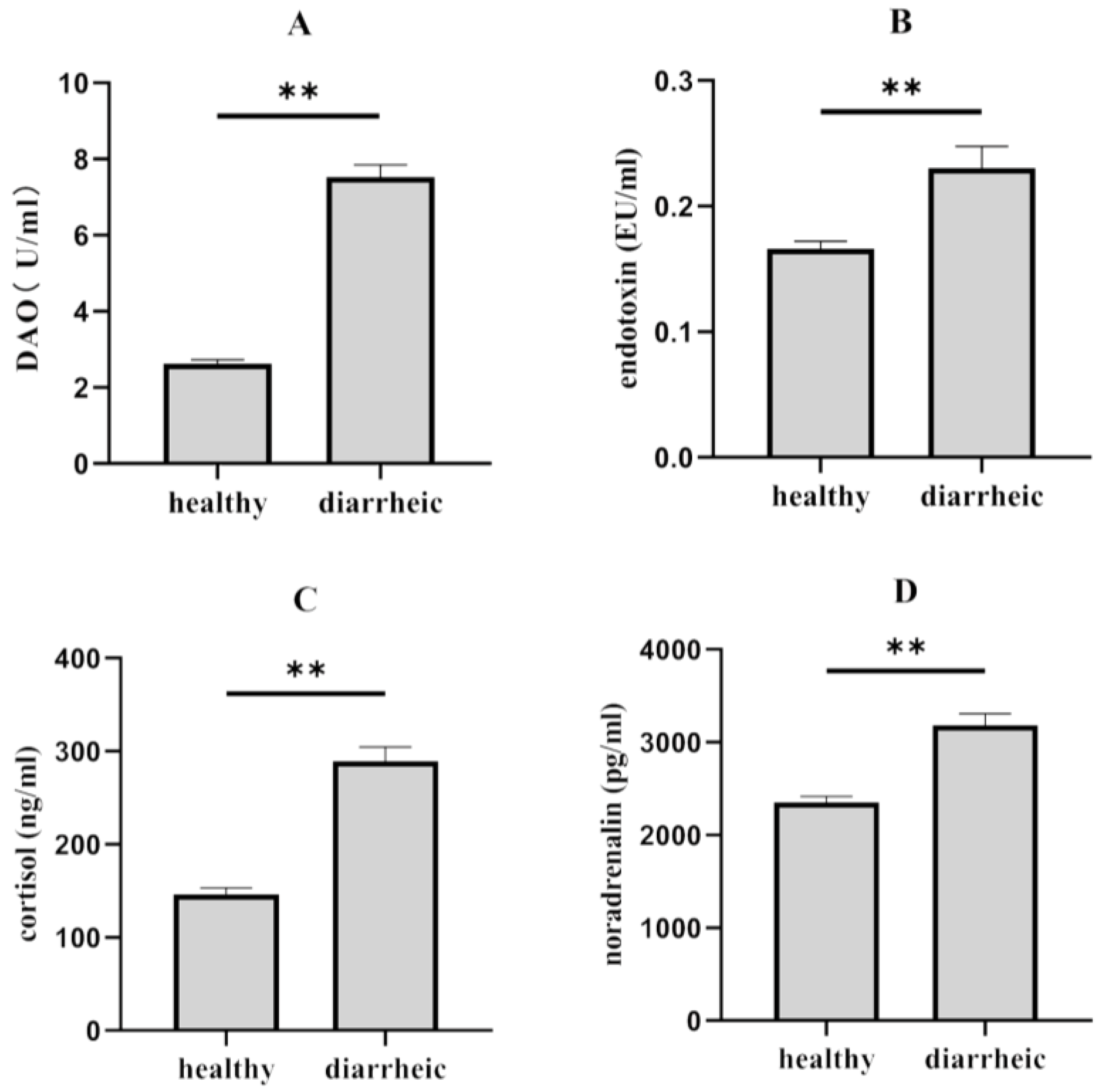
2.4. Plasma Biochemical Parameters Analysis
2.5. 16S rRNA Gene Sequencing and Sequencing Data Analysis
2.6. Metabolites Extraction for UHPLC-MS Analysis
2.7. Statistical Analysis
3. Results
3.1. Differences in Morphological and Plasma Physiological between Diarrheic and Healthy Piglets
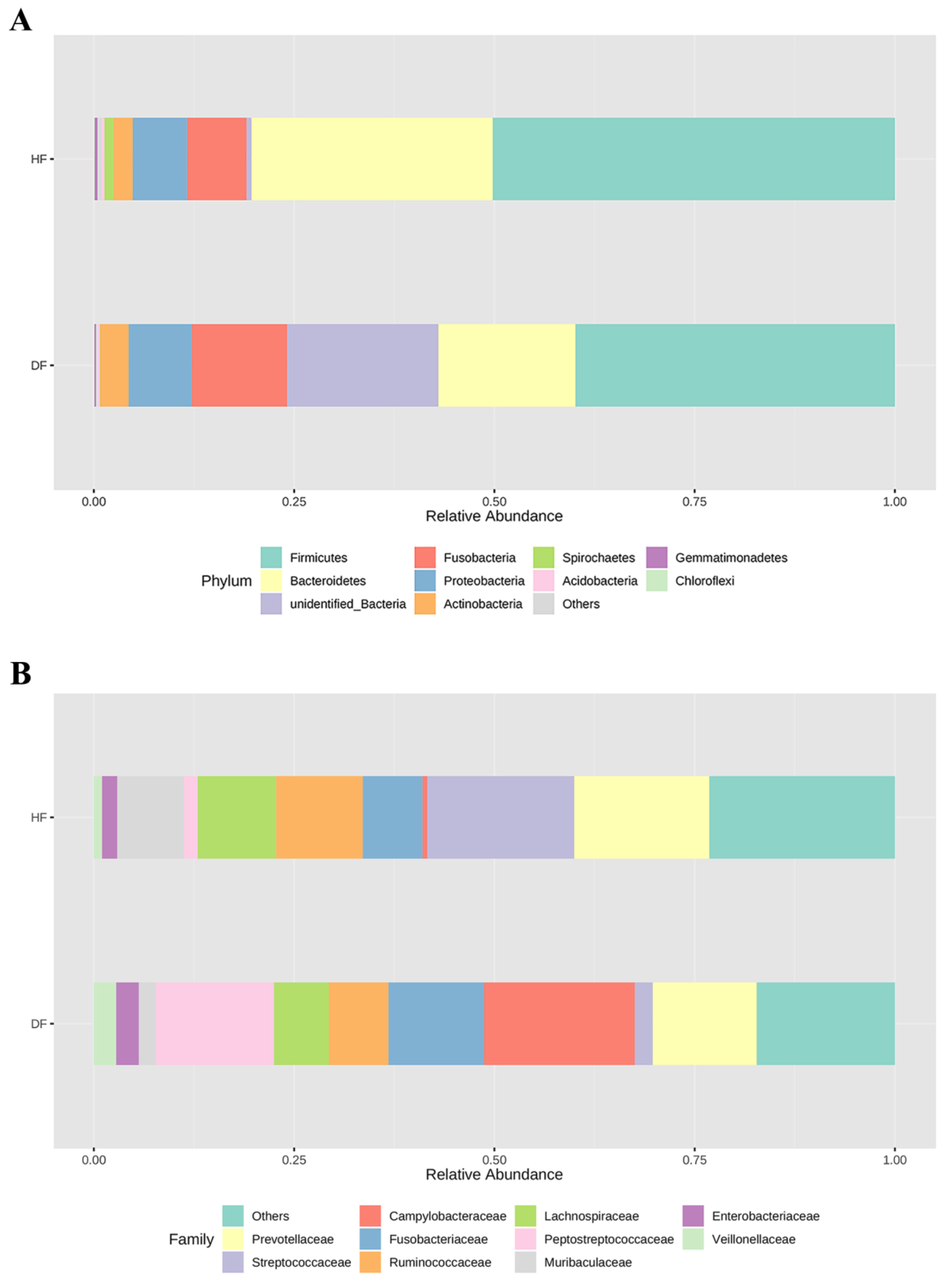
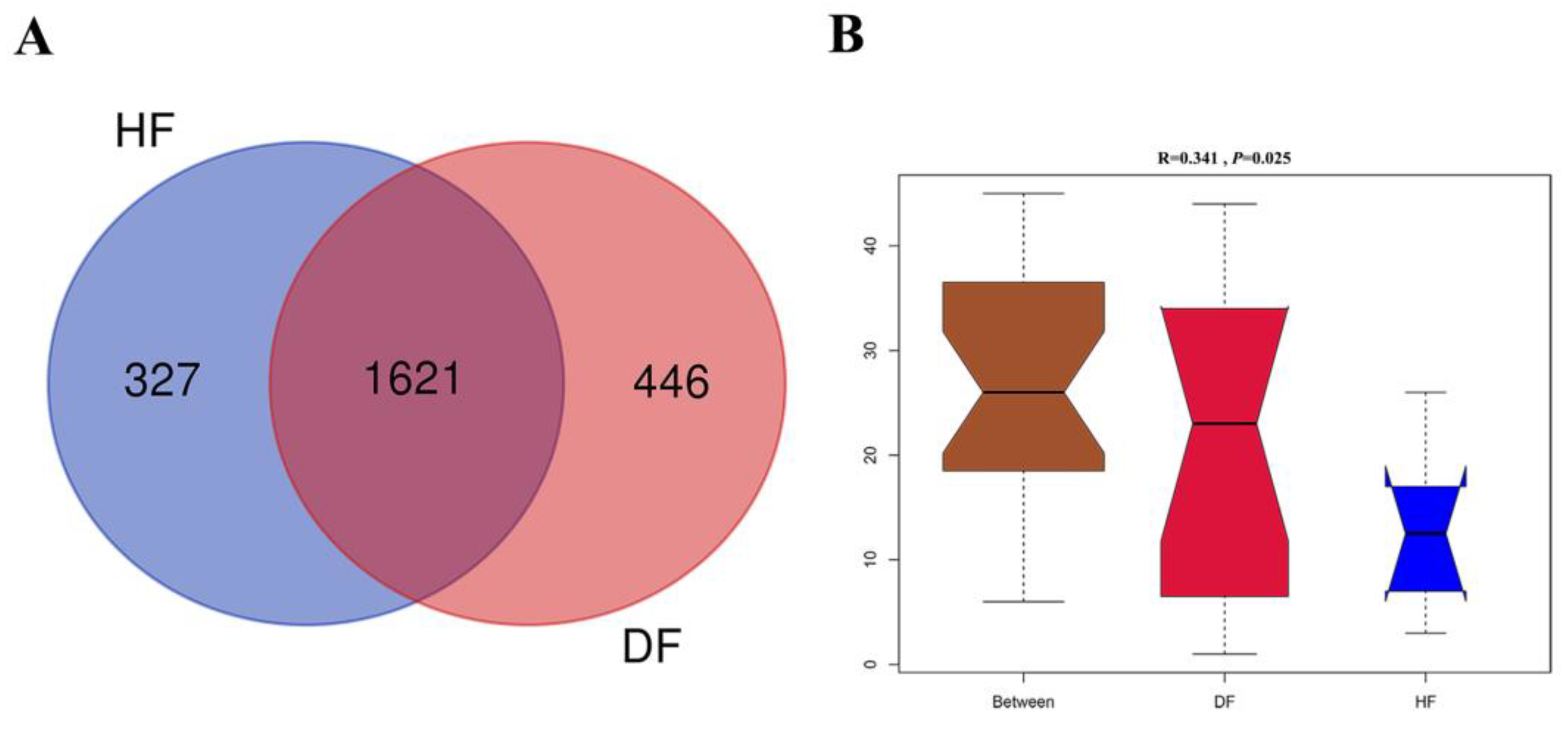
3.2. 16S rRNA Gene Profile in Diarrheic and Healthy Piglets
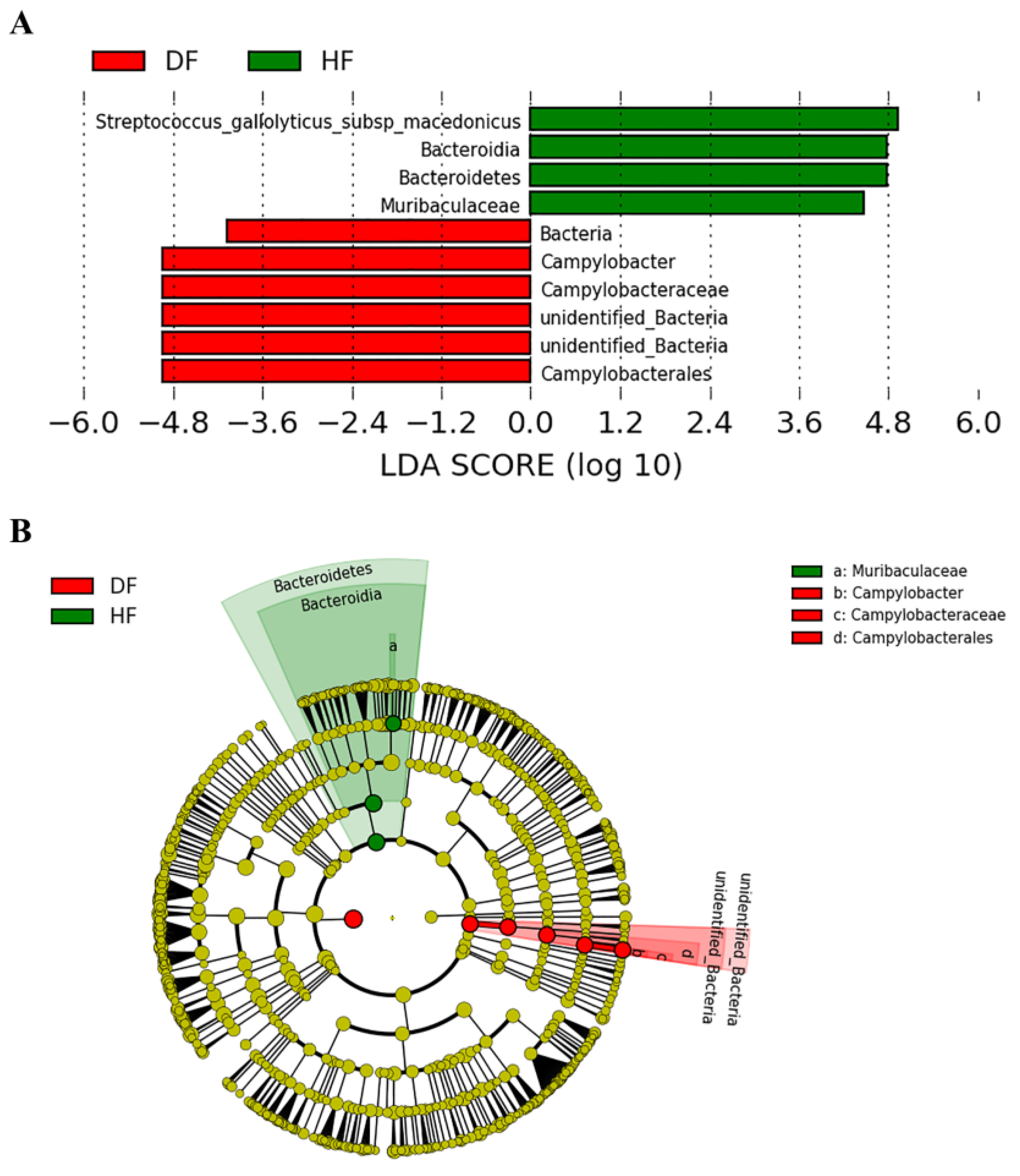
3.3. Differences of Gut Microbial Diversity and Composition in Diarrheic and Healthy Piglets
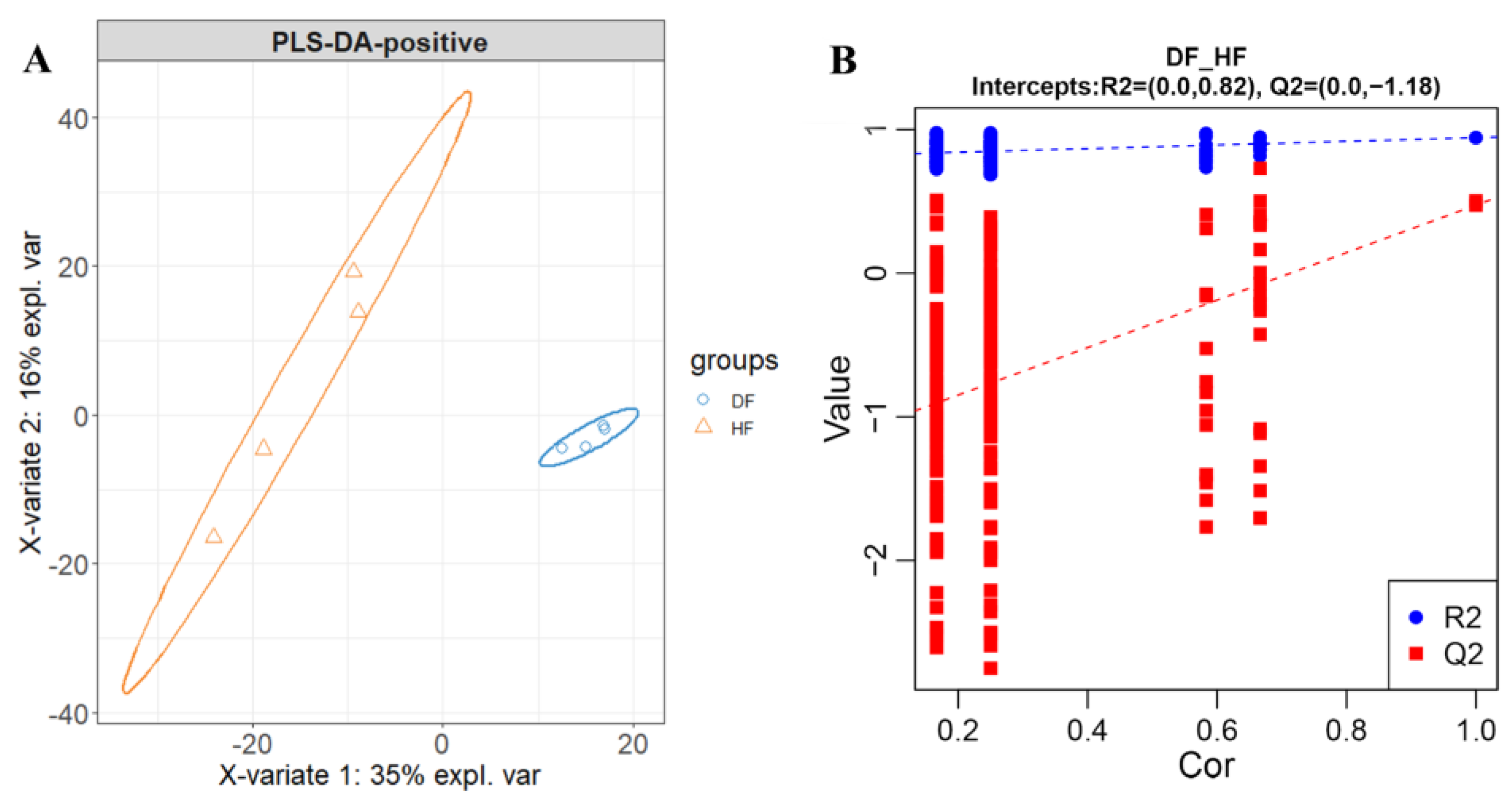
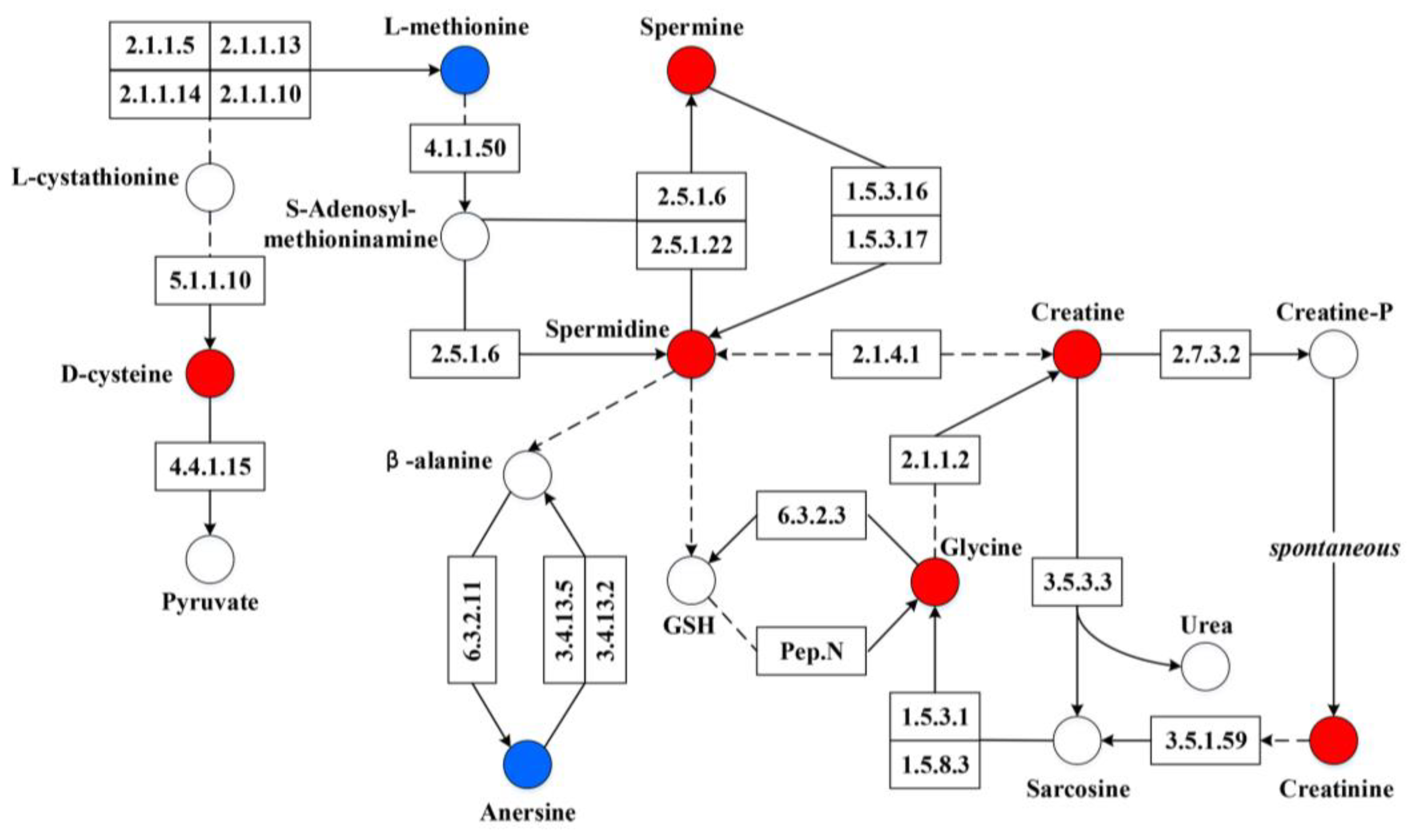
3.4. Metabolic Differences of Fecal Microbiota between Diarrheic and Healthy Piglets
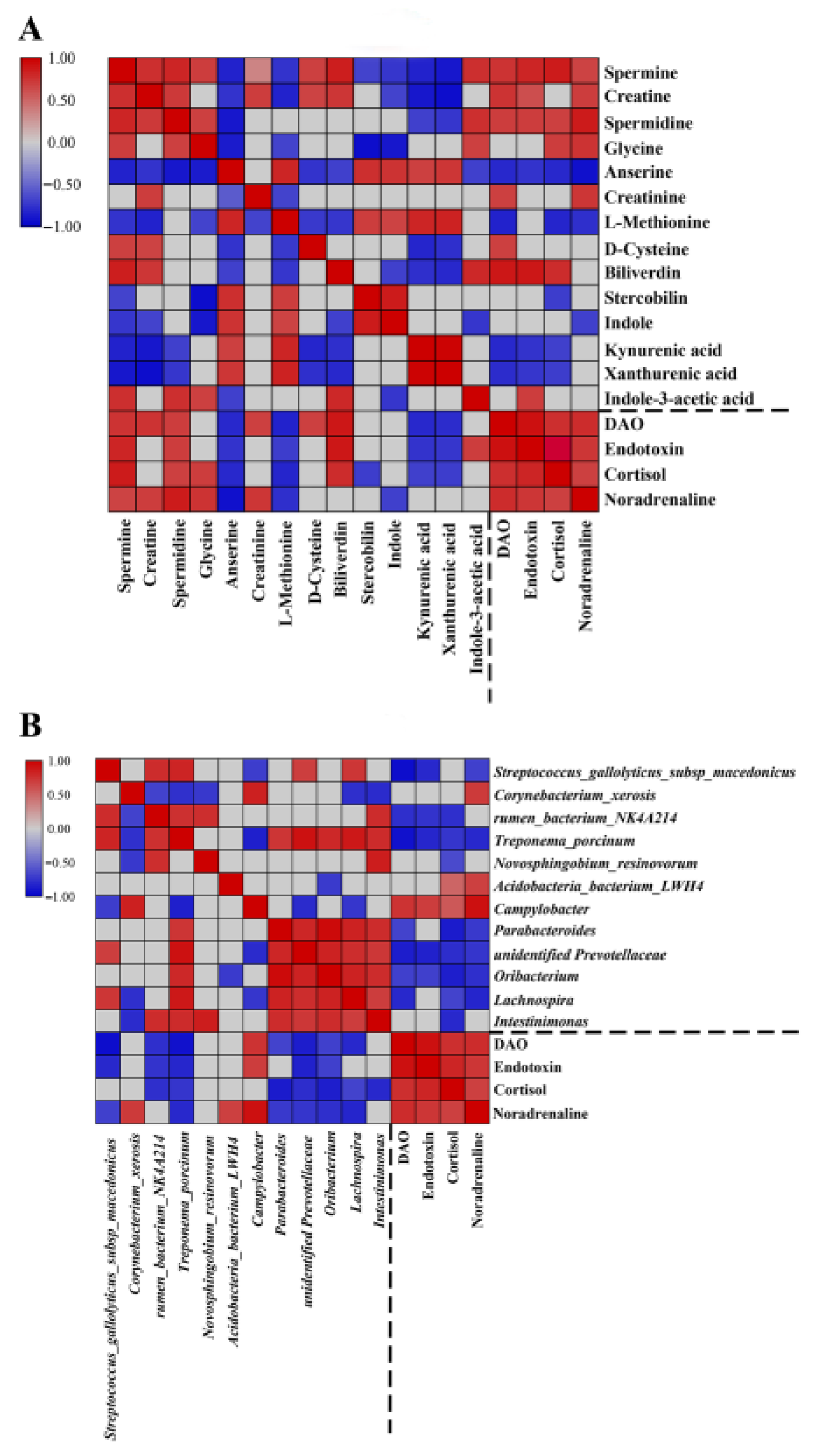
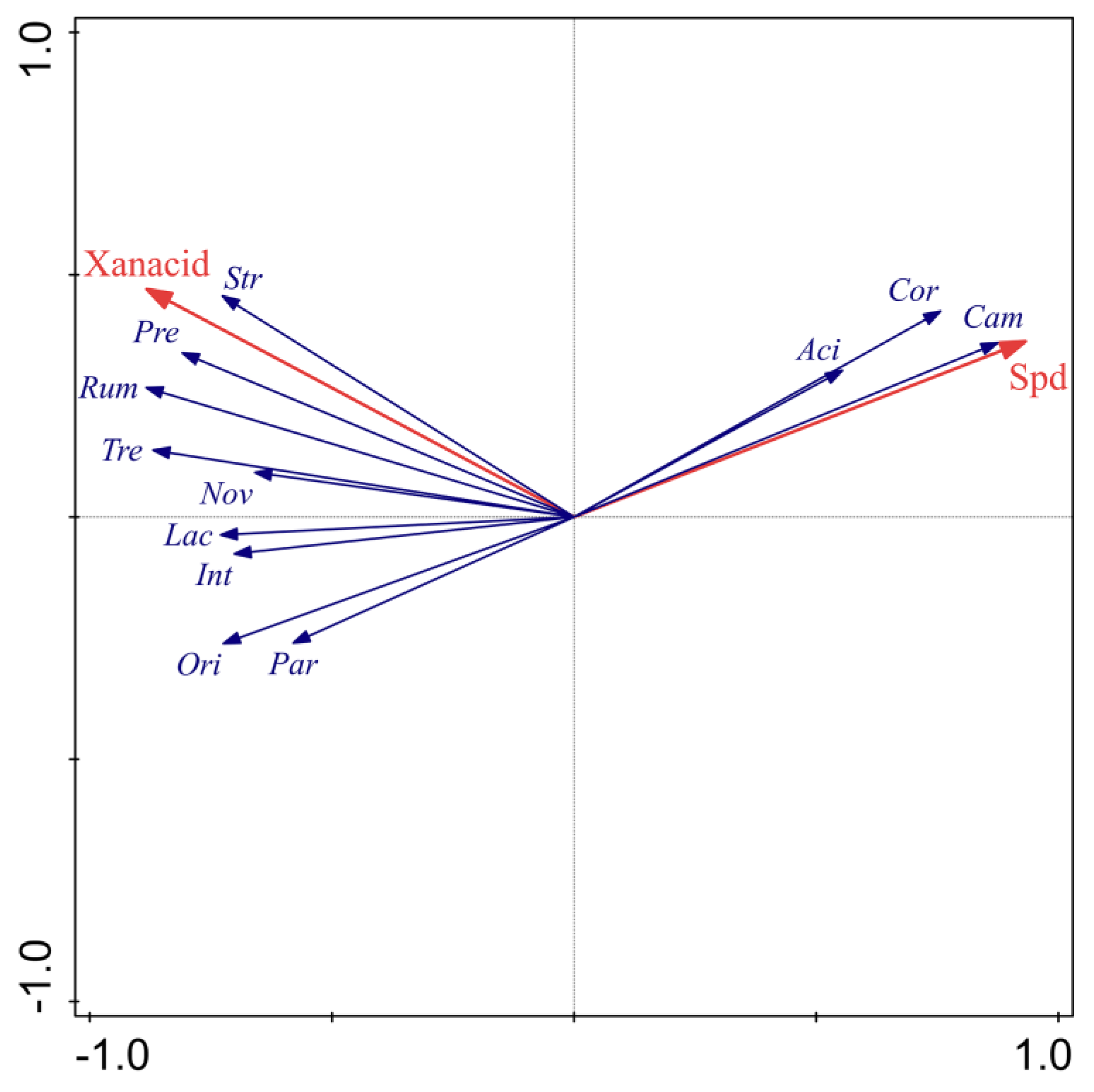
3.5. Correlation of the Fecal Microbiota, Plasma Physiological Parameters, and Fecal Metabolites in Diarrheic Piglets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morris, R.; Davies, P.; Lawton, D. Evolution of diseases in the world’s pig industry. In Proceedings of the 17th International Pig Veterinary Society Congress, Ames, IA, USA, 2–5 June 2002; pp. 1–10. [Google Scholar]
- Hermann-Bank, M.L. Characterization of the Bacterial Gut Microbiota in New Neonatal Porcine Diarrhoea. Ph.D. Thesis, Technical University of Denmark, Lyngby, Denmark, 2014. [Google Scholar]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Huang, X.; Zhao, S.; Sun, W.; Yan, Z.; Wang, P.; Li, S.; Huang, W.; Zhang, S.; Liu, L.; et al. Structure and Function of the Fecal Microbiota in Diarrheic Neonatal Piglets. Front. Microbiol. 2017, 8, 502. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.; Cole, J.R.; et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef]
- Gresse, R.; Chaucheyras-Durand, F.; Fleury, M.A.; Van de Wiele, T.; Forano, E.; Blanquet-Diot, S. Gut Microbiota Dysbiosis in Postweaning Piglets: Understanding the Keys to Health. Trends Microbiol. 2017, 25, 851–873. [Google Scholar] [CrossRef]
- Al-Asmakh, M.; Zadjali, F. Use of Germ-Free Animal Models in Microbiota-Related Research. J. Microbiol. Biotechnol. 2015, 25, 1583–1588. [Google Scholar] [CrossRef]
- Sanchez, B.; Delgado, S.; Blanco-Miguez, A.; Lourenco, A.; Gueimonde, M.; Margolles, A. Probiotics, gut microbiota, and their influence on host health and disease. Mol. Nutr. Food Res. 2017, 61, 1600240. [Google Scholar] [CrossRef]
- Baumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef]
- Pacheco, A.R.; Curtis, M.M.; Ritchie, J.M.; Munera, D.; Waldor, M.K.; Moreira, C.G.; Sperandio, V. Fucose sensing regulates bacterial intestinal colonization. Nature 2012, 492, 113–117. [Google Scholar] [CrossRef]
- Tran, T.H.T.; Everaert, N.; Bindelle, J. Review on the effects of potential prebiotics on controlling intestinal enteropathogens Salmonella and Escherichia coli in pig production. J. Anim. Physiol. Anim. Nutr. 2018, 102, 17–32. [Google Scholar] [CrossRef]
- Baidoo, E.E.K. Microbial Metabolomics: A General Overview. Methods Mol. Biol. 2019, 1859, 1–8. [Google Scholar] [PubMed]
- Sugiharto, S.; Hedemann, M.S.; Lauridsen, C. Plasma metabolomic profiles and immune responses of piglets after weaning and challenge with E. coli. J. Anim. Sci. Biotechnol. 2014, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, J.; Chen, S.; Zhao, Y.; Zeng, S.; Bin, P.; Zhang, D.; Tang, Z.; Zhu, G. Jejunal metabolic responses to Escherichia coli infection in piglets. Front. Microbiol. 2018, 9, 2465. [Google Scholar] [CrossRef] [PubMed]
- Jansson, J.; Willing, B.; Lucio, M.; Fekete, A.; Dicksved, J.; Halfvarson, J.; Tysk, C.; Schmitt-Kopplin, P. Metabolomics Reveals Metabolic Biomarkers of Crohn′s Disease. PLoS ONE 2009, 4, e6386. [Google Scholar] [CrossRef] [PubMed]
- Vaissaire, J.; Marcon, C.; Kobisch, M.; Menec, M.L.; Carnero, R.; Laroche, M.; Mirial, G. Importance of streptococcal disease (Streptococcus suis, group R) in France. Ann. Zootech. 1985, 34, 1985. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-y, Y.M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Nobumichi, H.; Masaharu, N.; Manabu, N.; Yoshifumi, H.; Koshiro, H.; Masakatsu, Y. Structural and functional alterations in the gut of parenterally or enterally fed rats. J. Surg. Res. 1989, 47, 129–133. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Gloeckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruening, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Li, S.; Xia, J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites 2020, 10, 186. [Google Scholar] [CrossRef]
- Morris, C. Multivariate Analysis of Ecological Data Using Canoco 5, 2nd ed.; Cambridge University Press: Cambridge, UK, 2015; p. 32. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Coker, A.O.; Isokpehi, R.D.; Thomas, B.N.; Amisu, K.O.; Obi, C.L. Human campylobacteriosis in developing countries. Emerg. Infect. Dis. 2002, 8, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Kirk, M.D.; Pires, S.M.; Black, R.E.; Caipo, M.; Crump, J.A.; Devleesschauwer, B.; Doepfer, D.; Fazil, A.; Fischer-Walker, C.L.; Hald, T.; et al. World Health Organization Estimates of the Global and Regional Disease Burden of 22 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med. 2015, 12, e1001940. [Google Scholar]
- Burrough, E.; Terhorst, S.; Sahin, O.; Zhang, Q. Prevalence of Campylobacter spp. relative to other enteric pathogens in grow-finish pigs with diarrhea. Anaerobe 2013, 22, 111–114. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Wen, Z.; Jiang, X.; Ma, X.; Han, X. Weaning Stress Perturbs Gut Microbiome and Its Metabolic Profile in Piglets. Sci. Rep. 2018, 8, 18068. [Google Scholar] [CrossRef]
- Robinson, I.M.; Whipp, S.C.; Bucklin, J.A.; Allison, M.J. Characterization of predominant bacteria from the colons of normal and dysenteric pigs. Appl. Environ. Microbiol. 1984, 48, 964–969. [Google Scholar] [CrossRef]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Suchodolski, J.S.; Markel, M.E.; Garcia-Mazcorro, J.F.; Unterer, S.; Heilmann, R.M.; Dowd, S.E.; Kachroo, P.; Ivanov, I.; Minamoto, Y.; Dillman, E.M.; et al. The Fecal Microbiome in Dogs with Acute Diarrhea and Idiopathic Inflammatory Bowel Disease. PLoS ONE 2012, 7, e51907. [Google Scholar] [CrossRef]
- Leser, T.D.; Amenuvor, J.Z.; Jensen, T.K.; Lindecrona, R.H.; Boye, M.; Moller, K. Culture-independent analysis of gut bacteria: The pig gastrointestinal tract microbiota revisited. Appl. Environ. Microbiol. 2002, 68, 673–690. [Google Scholar] [CrossRef]
- Pieterse, R.; Todorov, S.D.; Dicks, L.M.T. Bacteriocin ST91KM, produced by Streptococcus gallolyticus subsp. macedonicus ST91KM, is a narrow-spectrum peptide active against bacteria associated with mastitis in dairy cattle. Can. J. Microbiol. 2008, 54, 525–531. [Google Scholar]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rao, J.N.; Zou, T.; Liu, L.; Marasa, B.S.; Xiao, L.; Zeng, X.; Turner, D.J.; Wang, J.-Y. Polyamines are required for expression of Toll-like receptor 2 modulating intestinal epithelial barrier integrity. Am. J. Physiol.—Gastrointest. Liver Physiol. 2007, 293, G568–G576. [Google Scholar] [CrossRef] [PubMed]
- Hanfrey, C.C.; Pearson, B.M.; Hazeldine, S.; Lee, J.; Gaskin, D.J.; Woster, P.M.; Phillips, M.A.; Michael, A.J. Alternative Spermidine Biosynthetic Route Is Critical for Growth of Campylobacter jejuni and Is the Dominant Polyamine Pathway in Human Gut Microbiota. J. Biol. Chem. 2011, 286, 43301–43312. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, X.; Nie, K.; Xu, Y.; Zhang, H.; Xie, F.; Xu, L.; Zhang, Z.; Ding, Y.; Yin, Z.; Zhang, X. Fecal Microbial Structure and Metabolic Profile in Post-Weaning Diarrheic Piglets. Genes 2023, 14, 1166. https://doi.org/10.3390/genes14061166
Zheng X, Nie K, Xu Y, Zhang H, Xie F, Xu L, Zhang Z, Ding Y, Yin Z, Zhang X. Fecal Microbial Structure and Metabolic Profile in Post-Weaning Diarrheic Piglets. Genes. 2023; 14(6):1166. https://doi.org/10.3390/genes14061166
Chicago/Turabian StyleZheng, Xianrui, Ke Nie, Yiliang Xu, Huibin Zhang, Fan Xie, Liming Xu, Zhiyong Zhang, Yueyun Ding, Zongjun Yin, and Xiaodong Zhang. 2023. "Fecal Microbial Structure and Metabolic Profile in Post-Weaning Diarrheic Piglets" Genes 14, no. 6: 1166. https://doi.org/10.3390/genes14061166
APA StyleZheng, X., Nie, K., Xu, Y., Zhang, H., Xie, F., Xu, L., Zhang, Z., Ding, Y., Yin, Z., & Zhang, X. (2023). Fecal Microbial Structure and Metabolic Profile in Post-Weaning Diarrheic Piglets. Genes, 14(6), 1166. https://doi.org/10.3390/genes14061166