
Integrative Genetic Approach Facilitates Precision Strategies for Acute Myocardial Infarction
 , ,
, , 
Abstract
:1. Introduction
2. Atherosclerosis and Lipid Metabolism
3. Coagulation Cascade
4. Endothelial Function
5. Myocyte Stability
6. Genetic Risk Stratification for Acute Myocardial Infarction
Polygenic Risk Scores and Utility of Genetic Scores for AMI
7. Gene Therapy for Acute Myocardial Infarction
8. Challenges and Future Directions for Genomics in Acute Myocardial Infarction
8.1. Risk Stratification
8.2. Gene Therapy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar] [CrossRef]
- Murdock, D.R.; Venner, E.; Muzny, D.M.; Metcalf, G.A.; Murugan, M.; Hadley, T.D.; Chander, V.; de Vries, P.S.; Jia, X.; Hussain, A.; et al. Genetic testing in ambulatory cardiology clinics reveals high rate of findings with clinical management implications. Genet. Med. 2021, 23, 2404–2414. [Google Scholar] [CrossRef]
- Krittanawong, C.; Khawaja, M.; Rosenson, R.S.; Amos, C.I.; Nambi, V.; Lavie, C.J.; Virani, S.S. Association of PCSK9 Variants with the Risk of Atherosclerotic Cardiovascular Disease and Variable Responses to PCSK9 Inhibitor Therapy. Curr. Probl. Cardiol. 2022, 47, 101043. [Google Scholar] [CrossRef]
- Do, R.; Project, N.E.S.; Stitziel, N.O.; Won, H.-H.; Jørgensen, A.B.; Duga, S.; Merlini, P.A.; Kiezun, A.; Farrall, M.; Goel, A.; et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015, 518, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Tirdea, C.; Hostiuc, S.; Moldovan, H.; Scafa-Udriste, A. Identification of Risk Genes Associated with Myocardial Infarction—Big Data Analysis and Literature Review. Int. J. Mol. Sci. 2022, 23, 15008. [Google Scholar] [CrossRef]
- Tajima, T.; Morita, H.; Ito, K.; Yamazaki, T.; Kubo, M.; Komuro, I.; Momozawa, Y. Blood lipid-related low-frequency variants in LDLR and PCSK9 are associated with onset age and risk of myocardial infarction in Japanese. Sci. Rep. 2018, 8, 8107. [Google Scholar] [CrossRef] [Green Version]
- Pan-Lizcano, R.; Mariñas-Pardo, L.; Núñez, L.; Rebollal-Leal, F.; López-Vázquez, D.; Pereira, A.; Molina-Nieto, A.; Calviño, R.; Vázquez-Rodríguez, J.M.; Hermida-Prieto, M. Rare Variants in Genes of the Cholesterol Pathway Are Present in 60% of Patients with Acute Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 16127. [Google Scholar] [CrossRef]
- Hubacek, J.A. Apolipoprotein A5 and triglyceridemia. Focus on the effects of the common variants. Clin. Chem. Lab. Med. 2005, 43, 897–902. [Google Scholar] [CrossRef]
- Ramakrishnan, L.; Sachdev, H.A.; Sharma, M.; Abraham, R.; Prakash, S.; Gupta, D.; Singh, Y.; Bhaskar, S.; Sinha, S.; Chandak, G.R.; et al. Relationship of APOA5, PPARgamma and HL gene variants with serial changes in childhood body mass index and coronary artery disease risk factors in young adulthood. Lipids Health Dis. 2011, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lu, Z.; Zhang, J.; Yang, Y.; Shen, J.; Zhang, X.; Song, Y. The APOA5 rs662799 polymorphism is associated with dyslipidemia and the severity of coronary heart disease in Chinese women. Lipids Health Dis. 2016, 15, 170. [Google Scholar] [CrossRef] [Green Version]
- De Caterina, R.; Talmud, P.J.; Merlini, P.A.; Foco, L.; Pastorino, R.; Altshuler, D.; Mauri, F.; Peyvandi, F.; Lina, D.; Kathiresan, S.; et al. Strong association of the APOA5-1131T>C gene variant and early-onset acute myocardial infarction. Atherosclerosis 2011, 214, 397–403. [Google Scholar] [CrossRef]
- Shen, B.; Zhao, W.; Zheng, Y.; Chen, X.; Xu, W.; Li, S. An Association Study Identifies Two Single Nucleotide Polymorphisms on Chromosome 11q23.3 as a Risk Locus for Acute Myocardial Infarction in the Chinese Han Population. Clin. Lab. 2015, 61, 1609–1616. [Google Scholar] [CrossRef] [Green Version]
- Cesari, M.; Pahor, M.; Incalzi, R.A. REVIEW: Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [Green Version]
- Sobhan, M.R.; Mehdinezhad, M.; Moghimi, M.; Aghili, K.; Zare-Shehneh, M.; Neamatzadeh, H.; Miresmaeili, S.M. Plasminogen Activator Inhibitor-1 4G/5G polymorphism Contributes to Osteonecrosis of the Femoral Head Susceptibility: Evidence from a Systematic Review and Meta-Analysis. Arch. Bone Jt. Surg. 2018, 6, 468–477. [Google Scholar]
- French, D.; Hamilton, L.H.; Mattano, L.A.; Sather, H.N.; Devidas, M.; Nachman, J.B.; Relling, M.V. A PAI-1 (SERPINE1) polymorphism predicts osteonecrosis in children with acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Blood 2008, 111, 4496–4499. [Google Scholar] [CrossRef] [Green Version]
- Onalan, O.; Balta, G.; Oto, A.; Kabakci, G.; Tokgozoglu, L.; Aytemir, K.; Altay, C.; Gürgey, A.; Nazli, N. Plasminogen activator inhibitor-1 4G4G genotype is associated with myocardial infarction but not with stable coronary artery disease. J. Thromb. Thrombolysis 2008, 26, 211–217. [Google Scholar] [CrossRef]
- Nikolopoulos, G.K.; Bagos, P.G.; Tsangaris, I.; Tsiara, C.G.; Kopterides, P.; Vaiopoulos, A.; Kapsimali, V.; Bonovas, S.; Tsantes, A.E. The association between plasminogen activator inhibitor type 1 (PAI-1) levels, PAI-1 4G/5G polymorphism, and myocardial infarction: A Mendelian randomization meta-analysis. Clin. Chem. Lab. Med. 2014, 52, 937–950. [Google Scholar] [CrossRef]
- Morange, P.; Saut, N.; Alessi, M.-C.; Yudkin, J.; Margaglione, M.; Di Minno, G.; Hamsten, A.; Humphries, S.; Trégouët, D.-A.; Juhan-Vague, I. Association of Plasminogen Activator Inhibitor (PAI)-1 (SERPINE1) SNPs With Myocardial Infarction, Plasma PAI-1, and Metabolic Parameters: The HIFMECH study. Arter. Thromb. Vasc. Biol. 2007, 27, 2250–2257. [Google Scholar] [CrossRef] [Green Version]
- Parpugga, T.K.; Tatarunas, V.; Skipskis, V.; Kupstyte, N.; Zaliaduonyte-Peksiene, D.; Lesauskaitė, V. The Effect of PAI-1 4G/5G Polymorphism and Clinical Factors on Coronary Artery Occlusion in Myocardial Infarction. Dis. Markers 2015, 2015, 260101. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Verma, A.K.; Sagar, V.; Ranjan, R.; Sharma, R.; Tomar, P.; Bhatt, D.; Goyal, Y.; Alsahli, M.A.; Almatroudi, A.; et al. Genotype Variations and Association between PAI-1 Promoter Region (4G/5G and -844G/A) and Susceptibility to Acute Myocardial Infarction and Chronic Stable Angina. Cardiol. Res. Pr. 2021, 2021, 5551031. [Google Scholar] [CrossRef]
- El-Aziz, T.A.A.; Rezk, N.A. Relation of PAI-1 and TPA Genes Polymorphisms to Acute Myocardial Infarction and its Outcomes in Egyptian Patients. Cell Biochem. Biophys. 2015, 71, 227–234. [Google Scholar] [CrossRef]
- Boncoraglio, G.B.; Bodini, A.; Brambilla, C.; Carriero, M.R.; Ciusani, E.; Parati, E.A. An Effect of the PAI-1 4G/5G Polymorphism on Cholesterol Levels May Explain Conflicting Associations with Myocardial Infarction and Stroke. Cerebrovasc. Dis. 2006, 22, 191–195. [Google Scholar] [CrossRef]
- Kim, K.N.; Kim, K.M.; Kim, B.T.; Joo, N.S.; Choo, D.Y.; Lee, D.J. Relationship of plasminogen activator inhibitor 1 gene 4G/5G polymorphisms to hypertension in Korean women. Chin. Med. J. 2012, 125, 1249–1253. [Google Scholar]
- Meigs, J.B.; Dupuis, J.; Liu, C.; O’donnell, C.J.; Fox, C.S.; Kathiresan, S.; Gabriel, S.B.; Larson, M.G.; Yang, Q.; Herbert, A.G.; et al. PAI-1 Gene 4G/5G Polymorphism and Risk of Type 2 Diabetes in a Population-based Sample. Obesity 2006, 14, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Dong, P.; Yang, X.; Liu, Z. Plasminogen activator inhibitor-1 4G/5G polymorphism is associated with coronary artery disease risk: A meta-analysis. Int. J. Clin. Exp. Med. 2014, 7, 3777–3788. [Google Scholar]
- Rodríguez, I.; Coto, E.; Reguero, J.R.; González, P.; Andrés, V.; Lozano, I.; Martín, M.; Alvarez, V.; Morís, C. Role of the CDKN1A/p21, CDKN1C/p57, and CDKN2A/p16 Genes in the Risk of Atherosclerosis and Myocardial Infarction. Cell Cycle 2007, 6, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Yang, X. Identification of risk genes associated with myocardial infarction based on the recursive feature elimination algorithm and support vector machine classifier. Mol. Med. Rep. 2018, 17, 1555–1560. [Google Scholar] [CrossRef] [Green Version]
- Sumandea, C.A.; Garcia-Cazarin, M.L.; Bozio, C.H.; Sievert, G.A.; Balke, C.W.; Sumandea, M.P. Cardiac Troponin T, a Sarcomeric AKAP, Tethers Protein Kinase A at the Myofilaments. J. Biol. Chem. 2011, 286, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, J.; Drum, B.M.; Chen, Y.; Yin, H.; Guo, X.; Luckey, S.W.; Gilbert, M.L.; McKnight, G.S.; Scott, J.D.; et al. Loss of AKAP150 promotes pathological remodelling and heart failure propensity by disrupting calcium cycling and contractile reserve. Cardiovasc. Res. 2017, 113, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Lehnart, S.E.; Wehrens, X.H.T.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.-L.C.; Conti, M.; Marks, A.R. Phosphodiesterase 4D Deficiency in the Ryanodine-Receptor Complex Promotes Heart Failure and Arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Gulati, M.; Levy, P.D.; Mukherjee, D.; Amsterdam, E.; Bhatt, D.L.; Birtcher, K.K.; Blankstein, R.; Boyd, J.; Palmer, R.P.; Conejo, T.; et al. 2021 AHA/ACC/ASE/CHEST/SAEM/SCCT/SCMR Guideline for the Evaluation and Diagnosis of Chest Pain: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2021, 144, e368–e454. [Google Scholar]
- O’sullivan, J.W.; Raghavan, S.; Marquez-Luna, C.; Luzum, J.A.; Damrauer, S.M.; Ashley, E.A.; O’donnell, C.J.; Willer, C.J.; Natarajan, P. Polygenic Risk Scores for Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2022, 146, e93–e118. [Google Scholar] [CrossRef]
- Arnett, D.K.; Khera, A.; Blumenthal, R.S. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Part 1, Lifestyle and Behavioral Factors. JAMA Cardiol. 2019, 4, 1043–1044. [Google Scholar] [CrossRef]
- Mars, N.; Gen, F.; Koskela, J.T.; Ripatti, P.; Kiiskinen, T.T.J.; Havulinna, A.S.; Lindbohm, J.V.; Ahola-Olli, A.; Kurki, M.; Karjalainen, J.; et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat. Med. 2020, 26, 549–557. [Google Scholar] [CrossRef]
- Aragam, K.G.; Dobbyn, A.; Judy, R.; Chaffin, M.; Chaudhary, K.; Hindy, G.; Cagan, A.; Finneran, P.; Weng, L.-C.; Loos, R.J.; et al. Limitations of Contemporary Guidelines for Managing Patients at High Genetic Risk of Coronary Artery Disease. J. Am. Coll. Cardiol. 2020, 75, 2769–2780. [Google Scholar] [CrossRef]
- Elliott, J.; Bodinier, B.; Bond, T.A.; Chadeau-Hyam, M.; Evangelou, E.; Moons, K.G.M.; Dehghan, A.; Muller, D.C.; Elliott, P.; Tzoulaki, I. Predictive Accuracy of a Polygenic Risk Score–Enhanced Prediction Model vs a Clinical Risk Score for Coronary Artery Disease. JAMA 2020, 323, 636–645. [Google Scholar] [CrossRef]
- Riveros-Mckay, F.; Weale, M.E.; Moore, R.; Selzam, S.; Krapohl, E.; Sivley, R.M.; Tarran, W.A.; Sørensen, P.; Lachapelle, A.S.; Griffiths, J.A.; et al. Integrated Polygenic Tool Substantially Enhances Coronary Artery Disease Prediction. Circ. Genom. Precis. Med. 2021, 14, e003304. [Google Scholar] [CrossRef]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J. Am. Coll. Cardiol. 2018, 72, 1883–1893. [Google Scholar] [CrossRef]
- Manikpurage, H.D.; Eslami, A.; Perrot, N.; Li, Z.; Couture, C.; Mathieu, P.; Bossé, Y.; Arsenault, B.J.; Thériault, S. Polygenic Risk Score for Coronary Artery Disease Improves the Prediction of Early-Onset Myocardial Infarction and Mortality in Men. Circ. Genom. Precis. Med. 2021, 14, e003452. [Google Scholar] [CrossRef]
- Brautbar, A.; Pompeii, L.A.; Dehghan, A.; Ngwa, J.S.; Nambi, V.; Virani, S.S.; Rivadeneira, F.; Uitterlinden, A.G.; Hofman, A.; Witteman, J.C.; et al. A genetic risk score based on direct associations with coronary heart disease improves coronary heart disease risk prediction in the Atherosclerosis Risk in Communities (ARIC), but not in the Rotterdam and Framingham Offspring, Studies. Atherosclerosis 2012, 223, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Bhak, Y.; Jeon, Y.; Jeon, S.; Yoon, C.; Kim, M.; Blazyte, A.; Kim, Y.; Kang, Y.; Kim, C.; Lee, S.Y.; et al. Polygenic risk score validation using Korean genomes of 265 early-onset acute myocardial infarction patients and 636 healthy controls. PLoS ONE 2021, 16, e0246538. [Google Scholar] [CrossRef]
- Levin, M.G.; Rader, D.J. Polygenic Risk Scores and Coronary Artery Disease: Ready for Prime Time? Circulation 2020, 141, 637–640. [Google Scholar] [CrossRef]
- Williams, P.D.; Ranjzad, P.; Kakar, S.J.; Kingston, P.A. Development of Viral Vectors for Use in Cardiovascular Gene Therapy. Viruses 2010, 2, 334–371. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yu, L.; Zhou, A.; Liu, J.; Wang, K.; Luo, Y.; Wang, F. Non-Viral Gene Delivery Systems for Treatment of Myocardial Infarction: Targeting Strategies and Cardiac Cell Modulation. Pharmaceutics 2021, 13, 1520. [Google Scholar] [CrossRef]
- Janavel, G.V.; Crottogini, A.; Meckert, P.C.; Cuniberti, L.; Mele, A.; Papouchado, M.; Fernández, N.; Bercovich, A.; Criscuolo, M.; Melo, C.; et al. Plasmid-mediated VEGF gene transfer induces cardiomyogenesis and reduces myocardial infarct size in sheep. Gene Ther. 2006, 13, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Pajula, J.; Lähteenvuo, J.; Lähteenvuo, M.; Honkonen, K.; Halonen, P. Adenoviral VEGF-D(DeltaN DeltaC) gene therapy for myocardial ischemia. Front. Bioeng. Biotechnol. 2022, 10, 999226. [Google Scholar] [CrossRef]
- Hao, X.; Månsson-Broberg, A.; Grinnemo, K.-H.; Siddiqui, A.J.; Dellgren, G.; Brodin, L.; Sylvén, C. Myocardial angiogenesis after plasmid or adenoviral VEGF-A165 gene transfer in rat myocardial infarction model. Cardiovasc. Res. 2007, 73, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Penn, M.S. Importance of the SDF-1: CXCR4 Axis in Myocardial Repair. Circ. Res. 2009, 104, 1133–1135. [Google Scholar] [CrossRef] [Green Version]
- Kondo, I.; Ohmori, K.; Oshita, A.; Takeuchi, H.; Fuke, S.; Shinomiya, K.; Noma, T.; Namba, T.; Kohno, M. Treatment of Acute Myocardial Infarction by Hepatocyte Growth Factor Gene Transfer: The First Demonstration of Myocardial Transfer of a “Functional” Gene Using Ultrasonic Microbubble Destruction. J. Am. Coll. Cardiol. 2004, 44, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Kim, D.; Cho, B.; Byun, J.; Kim, Y.S.; Ahn, Y.; Hur, J.; Oh, Y.-K.; Kim, J. In vivo therapeutic genome editing via CRISPR/Cas9 magnetoplexes for myocardial infarction. Biomaterials 2022, 281, 121327. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Meng, D.; Han, S.; Jeong, I.S.; Kim, S.W. Interleukin 10-Secreting MSCs via TALEN-Mediated Gene Editing Attenuates Left Ventricular Remodeling after Myocardial Infarction. Cell. Physiol. Biochem. 2019, 52, 728–741. [Google Scholar]
- Li, F.; Yang, Y.; Xue, C.; Tan, M.; Xu, L.; Gao, J.; Xu, L.; Zong, J.; Qian, W. Zinc Finger Protein ZBTB20 protects against cardiac remodelling post-myocardial infarction via ROS-TNFalpha/ASK1/JNK pathway regulation. J. Cell. Mol. Med. 2020, 24, 13383–13396. [Google Scholar] [CrossRef]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef]
- Chu, H.; Kohane, D.S.; Langer, R. RNA therapeutics—The potential treatment for myocardial infarction. Regen. Ther. 2016, 4, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Ku, S.H.; Lee, M.S.; Jeong, J.H.; Mok, H.; Choi, D.; Kim, S.H. Cardiac RNAi therapy using RAGE siRNA/deoxycholic acid-modified polyethylenimine complexes for myocardial infarction. Biomaterials 2014, 35, 7562–7573. [Google Scholar] [CrossRef]
- Santos, R.D.; Raal, F.J.; Catapano, A.L.; Witztum, J.L.; Steinhagen-Thiessen, E.; Tsimikas, S. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: Results of 4 phase III trials. Arter. Thromb. Vasc. Biol. 2015, 35, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Behbodikhah, J.; Ahmed, S.; Elyasi, A.; Kasselman, L.J.; De Leon, J.; Glass, A.D.; Reiss, A.B. Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target. Metabolites 2021, 11, 690. [Google Scholar] [CrossRef]
- Merki, E.; Graham, M.; Taleb, A.; Leibundgut, G.; Yang, X.; Miller, E.R.; Fu, W.; Mullick, A.E.; Lee, R.; Willeit, P.; et al. Antisense Oligonucleotide Lowers Plasma Levels of Apolipoprotein (a) and Lipoprotein (a) in Transgenic Mice. J. Am. Coll. Cardiol. 2011, 57, 1611–1621. [Google Scholar] [CrossRef] [Green Version]
- Krittanawong, C.; Johnson, K.W.; Choi, E.; Kaplin, S.; Venner, E.; Murugan, M.; Wang, Z.; Glicksberg, B.S.; Amos, C.I.; Schatz, M.C.; et al. Artificial Intelligence and Cardiovascular Genetics. Life 2022, 12, 279. [Google Scholar] [CrossRef]
- Krittanawong, C.; Johnson, K.W.; Glicksberg, B.S. Opportunities and challenges for artificial intelligence in clinical cardiovascular genetics. Trends Genet. 2021, 37, 780–783. [Google Scholar] [CrossRef]


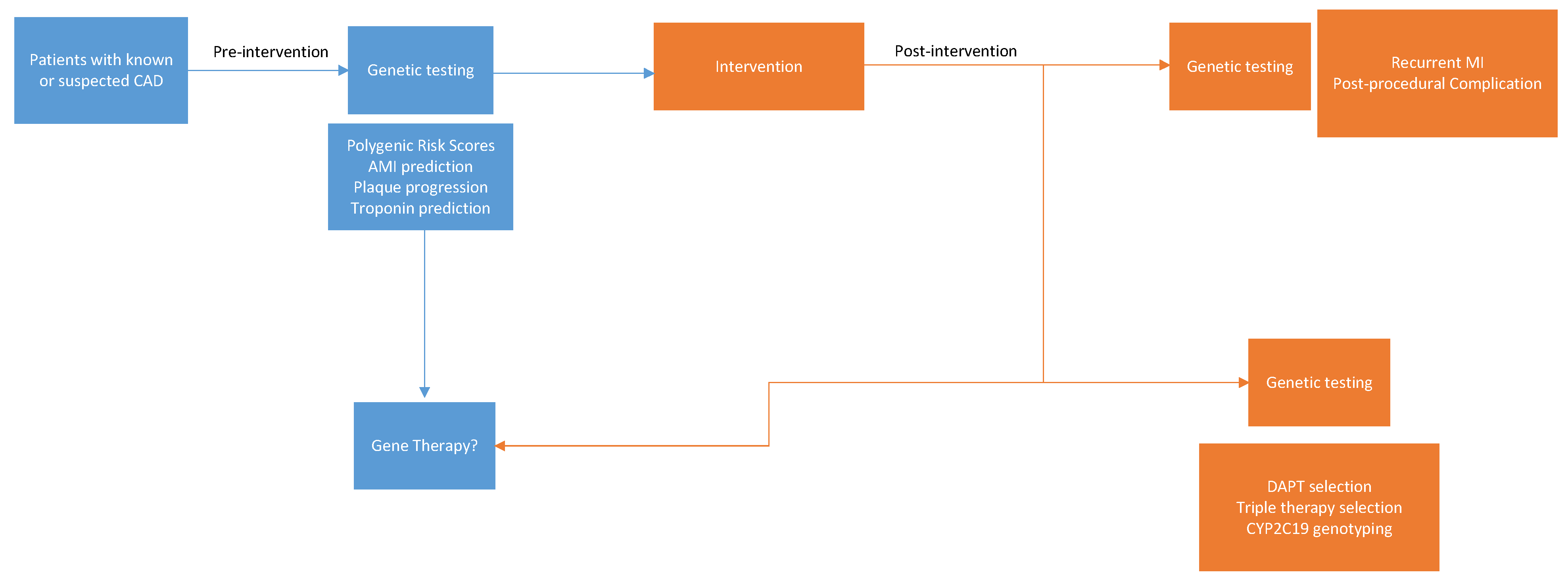{kind=link}
| Cellular Process | Gene | Normal Function of Protein |
|---|---|---|
| Lipid Metabolism | LDLR | Clearance of LDL from circulation via endocytosis |
| Lipid Metabolism | APO-A5 | Upregulation of lipoprotein lipase |
| Coagulation Cascade | PAI-1 | Inhibition of tissue plasminogen activator |
| Endothelial Function | CDKN1A | Regulation of vascular smooth muscle cell proliferation |
| Myocyte Stability | GLAR2 | Unknown |
| Myocyte Stability | AKAP | Regulation of cardiac contractility, remodeling and structural scaffolding |
| Therapy | Mechanism | Examples |
|---|---|---|
| Gene Transfer | Integrating new genetic material into a genome to treat or prevent disease via the use of viral and non-viral vectors | VEGF gene transfer to promote angiogenesis and improve blood supply to ischemic myocardium; |
| SDF-1 gene transfer enhances stem cell recruitment and homing to the injured heart to induce cardiac repair and regeneration; | ||
| HGF gene transfer promotes cardiomyocyte survival, angiogenesis, and tissue repair | ||
| Gene Editing | Altering the genome using CRISPR-Cas9, zinc finger nucleases, and transcription activator | CRISPR-Cas9 allows precise modification of the genome, enabling the correction of disease-causing genetic mutations associated with MI |
| Gene Silencing | Silencing genes via the use of siRNAs | SiRNAs to target pro-inflammatory cytokines to reduce inflammation and improve cardiac function; |
| Antisense oligonucleotides targeting genes involved in lipid metabolism, such as apolipoprotein-B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khawaja, M.; Siddiqui, R.; Virani, S.S.; Amos, C.I.; Bandyopadhyay, D.; Virk, H.U.H.; Alam, M.; Jneid, H.; Krittanawong, C. Integrative Genetic Approach Facilitates Precision Strategies for Acute Myocardial Infarction. Genes 2023, 14, 1340. https://doi.org/10.3390/genes14071340
Khawaja M, Siddiqui R, Virani SS, Amos CI, Bandyopadhyay D, Virk HUH, Alam M, Jneid H, Krittanawong C. Integrative Genetic Approach Facilitates Precision Strategies for Acute Myocardial Infarction. Genes. 2023; 14(7):1340. https://doi.org/10.3390/genes14071340
Chicago/Turabian StyleKhawaja, Muzamil, Rehma Siddiqui, Salim S. Virani, Christopher I. Amos, Dhrubajyoti Bandyopadhyay, Hafeez Ul Hassan Virk, Mahboob Alam, Hani Jneid, and Chayakrit Krittanawong. 2023. "Integrative Genetic Approach Facilitates Precision Strategies for Acute Myocardial Infarction" Genes 14, no. 7: 1340. https://doi.org/10.3390/genes14071340
APA StyleKhawaja, M., Siddiqui, R., Virani, S. S., Amos, C. I., Bandyopadhyay, D., Virk, H. U. H., Alam, M., Jneid, H., & Krittanawong, C. (2023). Integrative Genetic Approach Facilitates Precision Strategies for Acute Myocardial Infarction. Genes, 14(7), 1340. https://doi.org/10.3390/genes14071340