
Nutrient-Sensing Ghrelin Receptor in Macrophages Modulates Bisphenol A-Induced Intestinal Inflammation in Mice

 ,
, _Shen.jpg) , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Flow Cytometry Analysis
2.3. Histology
2.4. Generation of Ghsr Deletion Mutant in RAW264.7 Cells
2.5. Macrophage Culture and Treatment
2.6. Real-Time Quantitative PCR
2.7. Statistical Analysis
3. Results
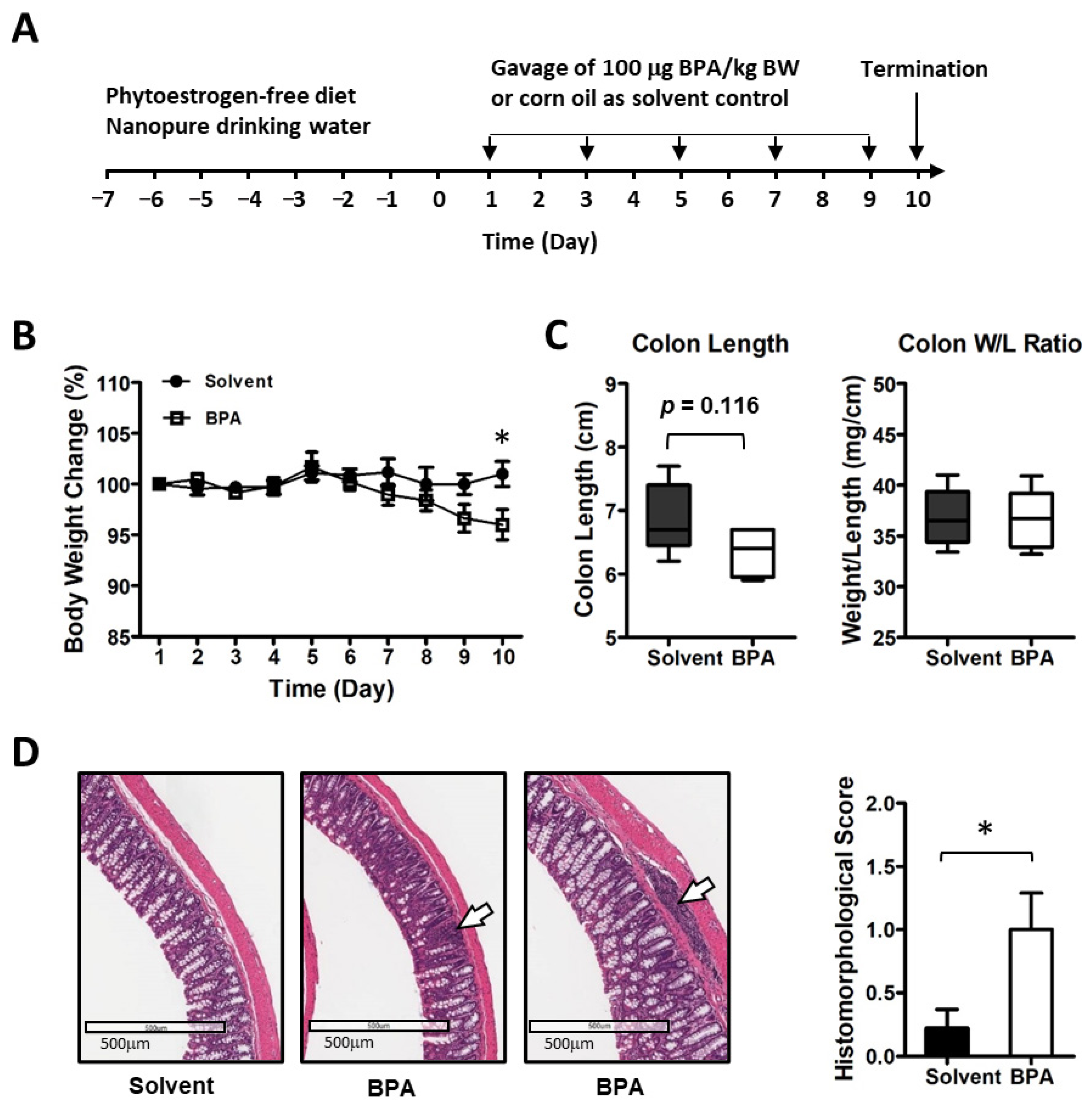
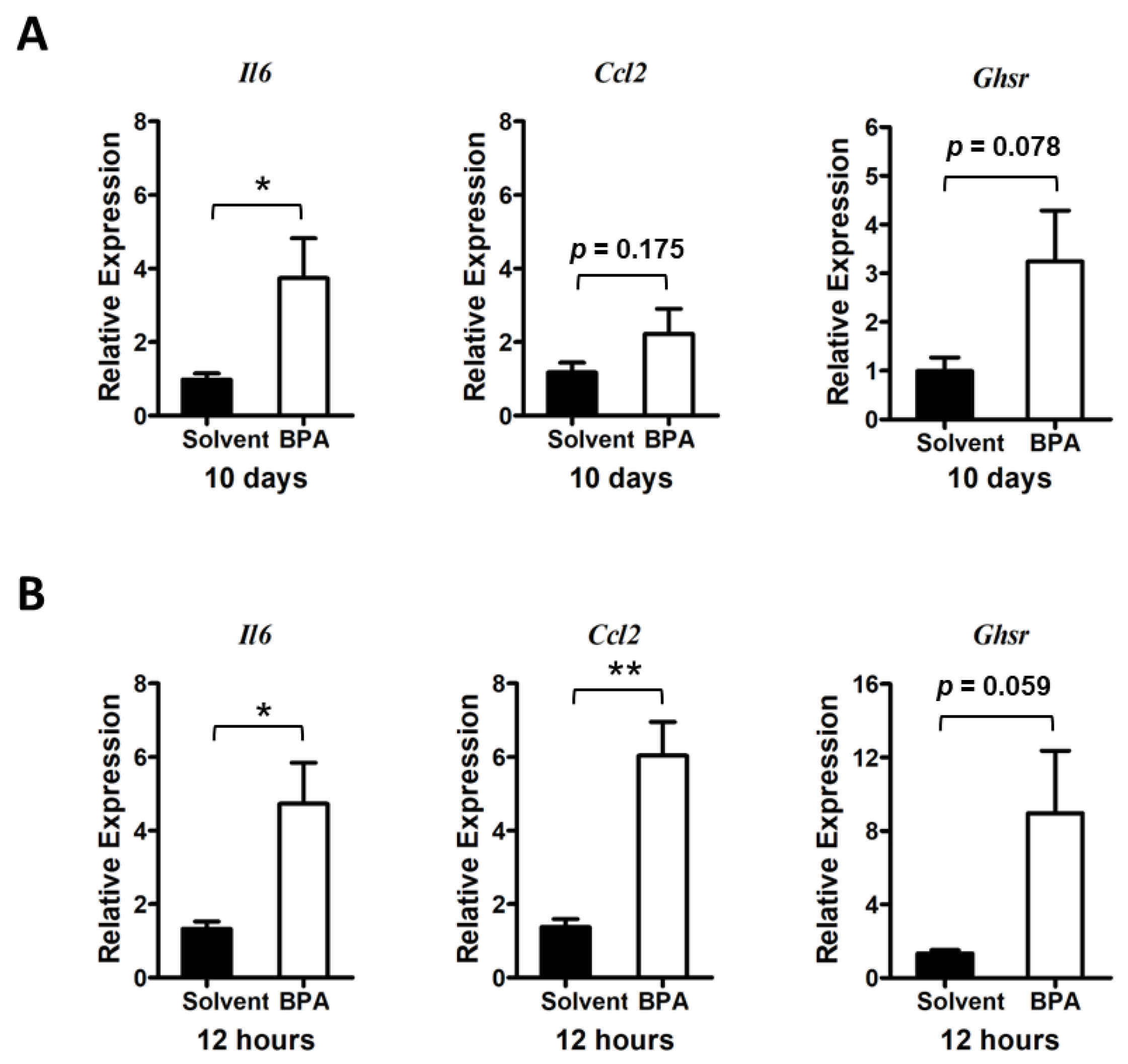
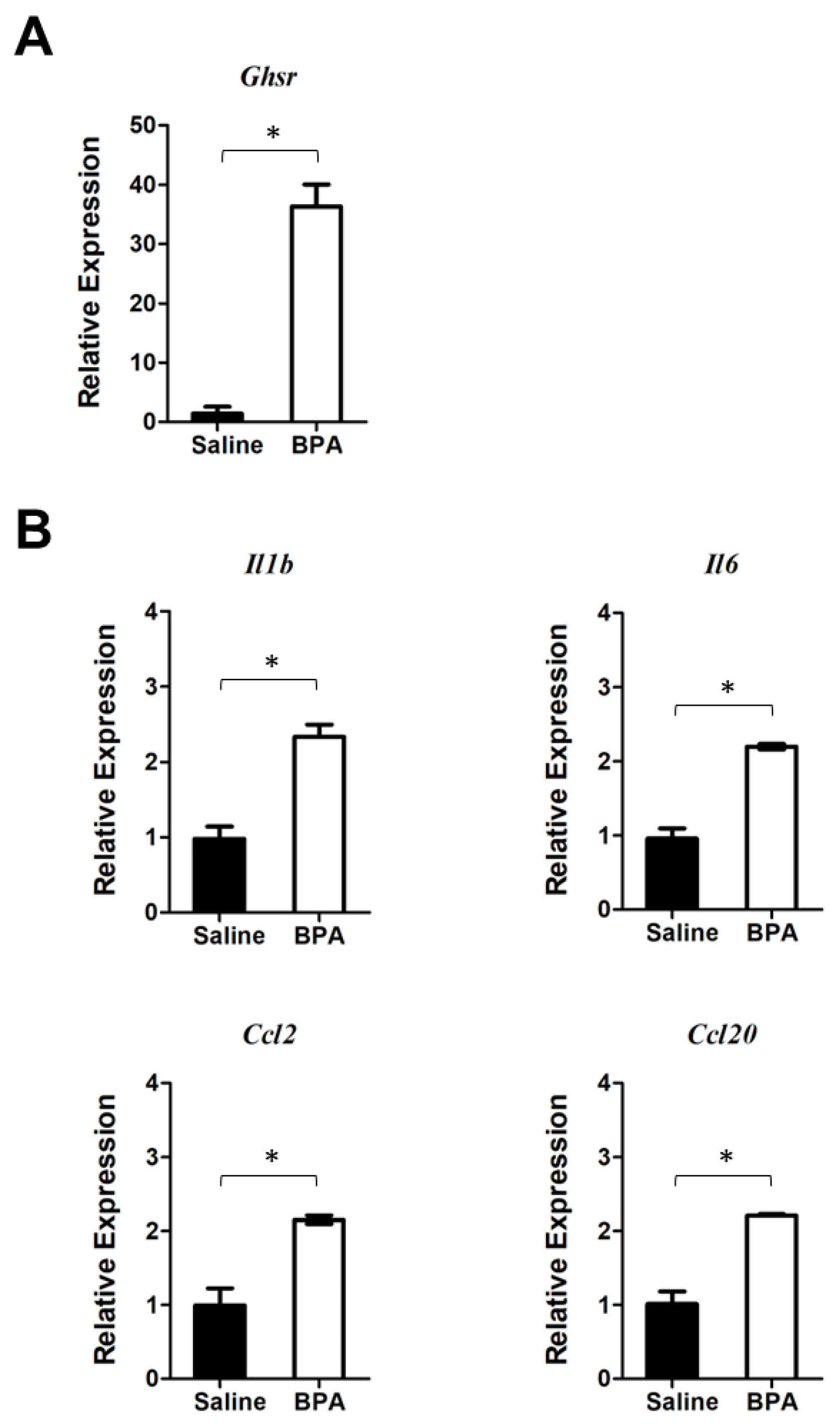
3.1. BPA Induces Inflammatory Responses in Colonic Mucosa of Mice
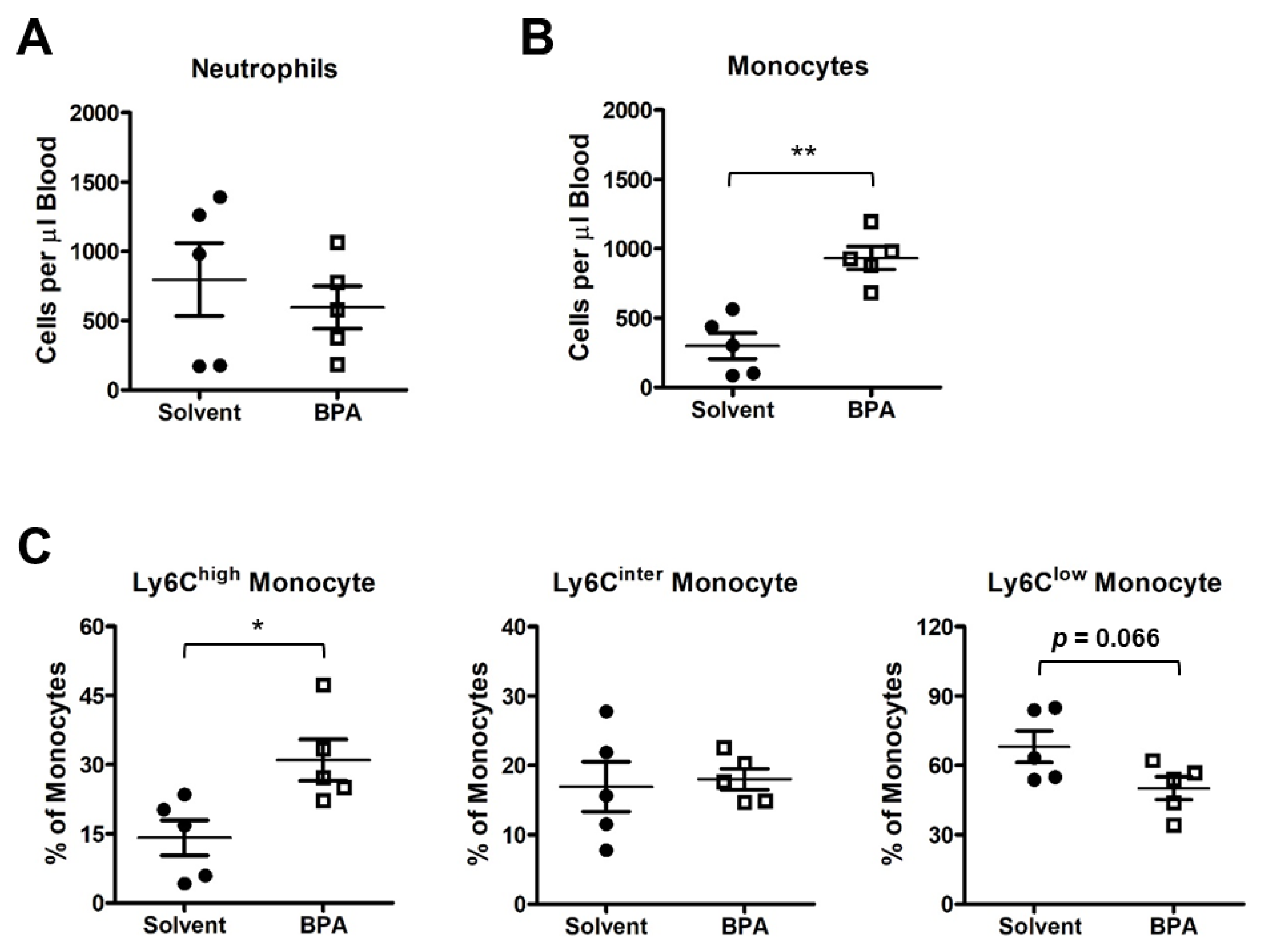
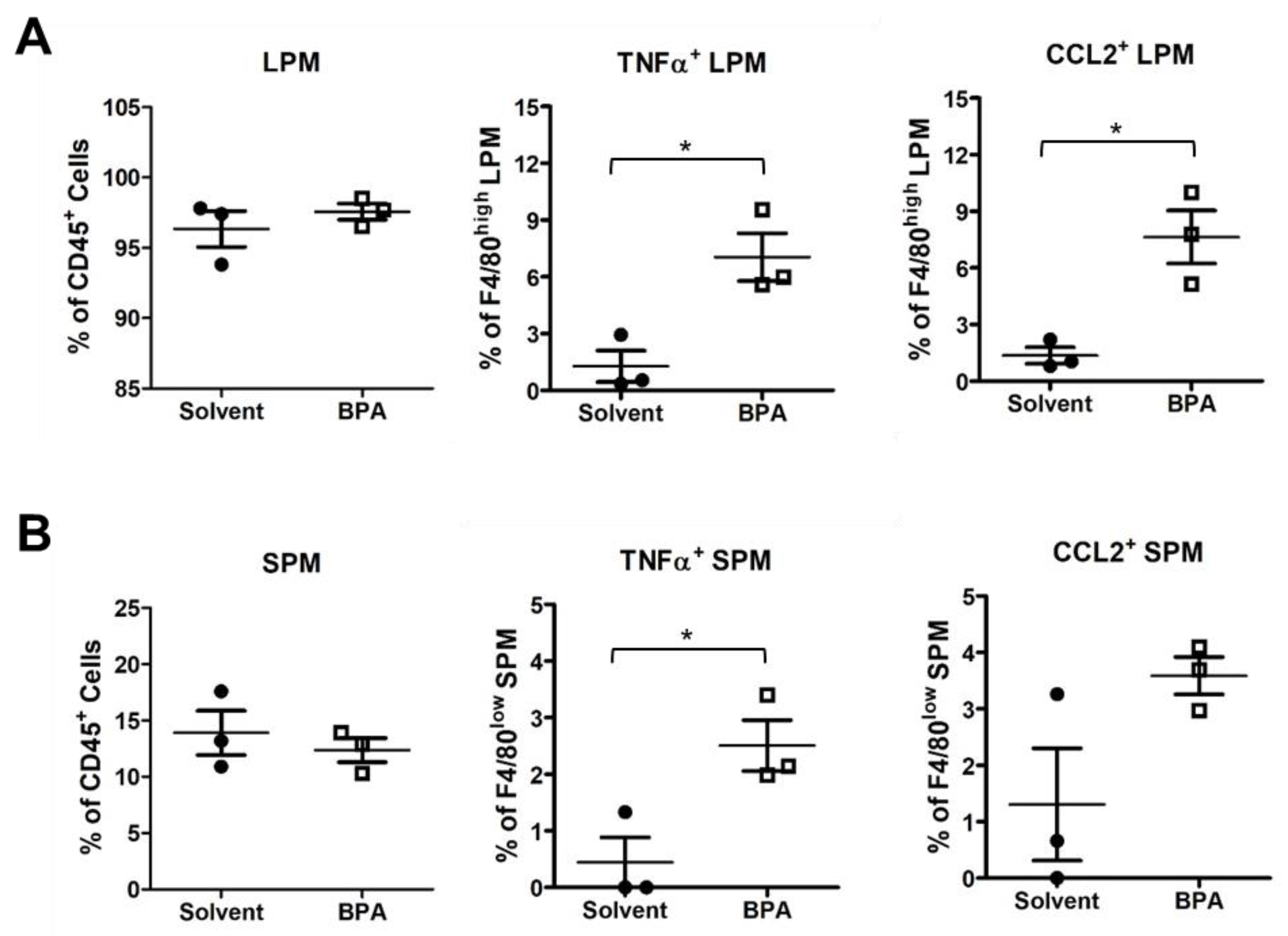
3.2. BPA Exposure Induces Systemic Activation of Monocytes and Macrophages
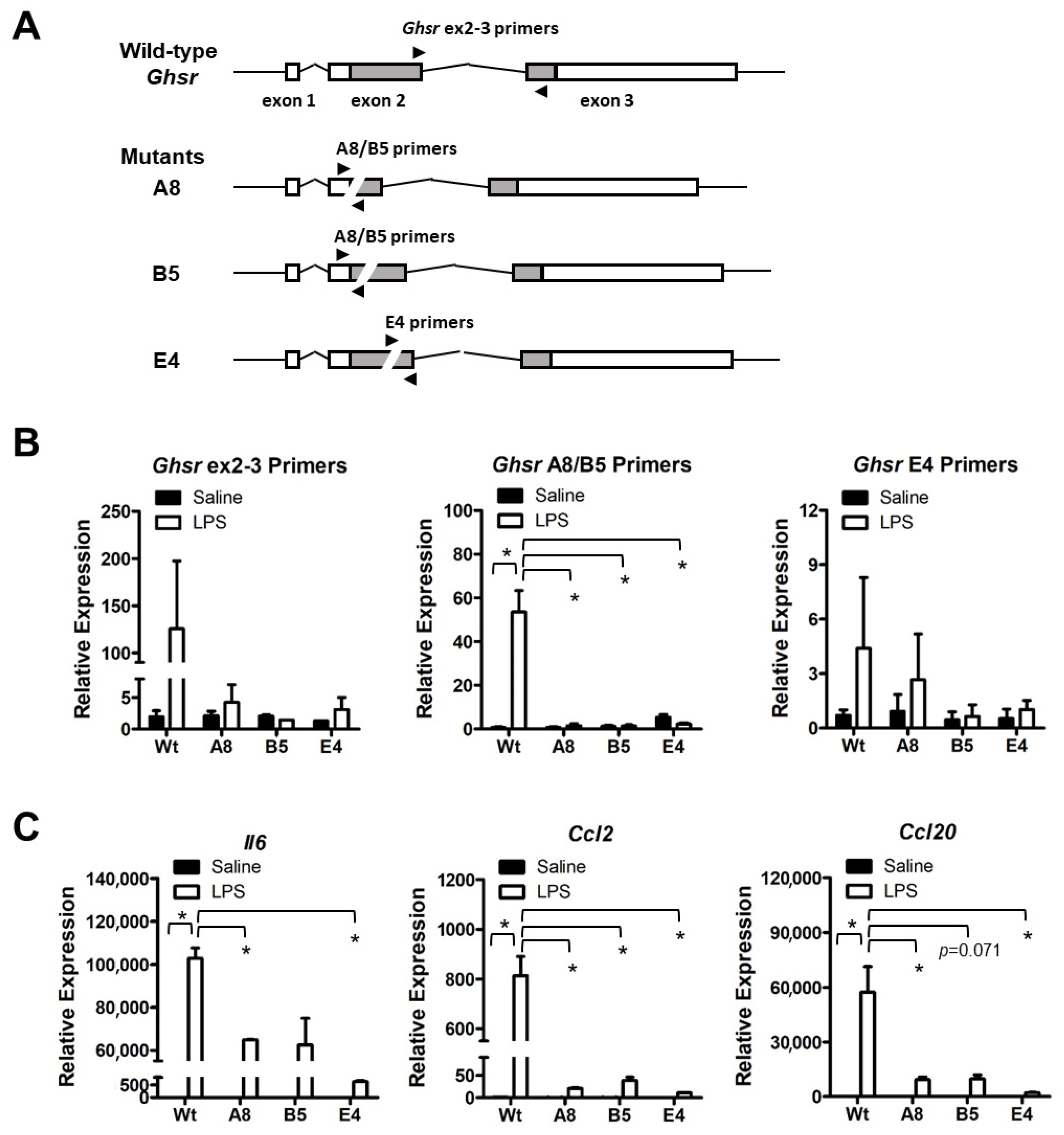
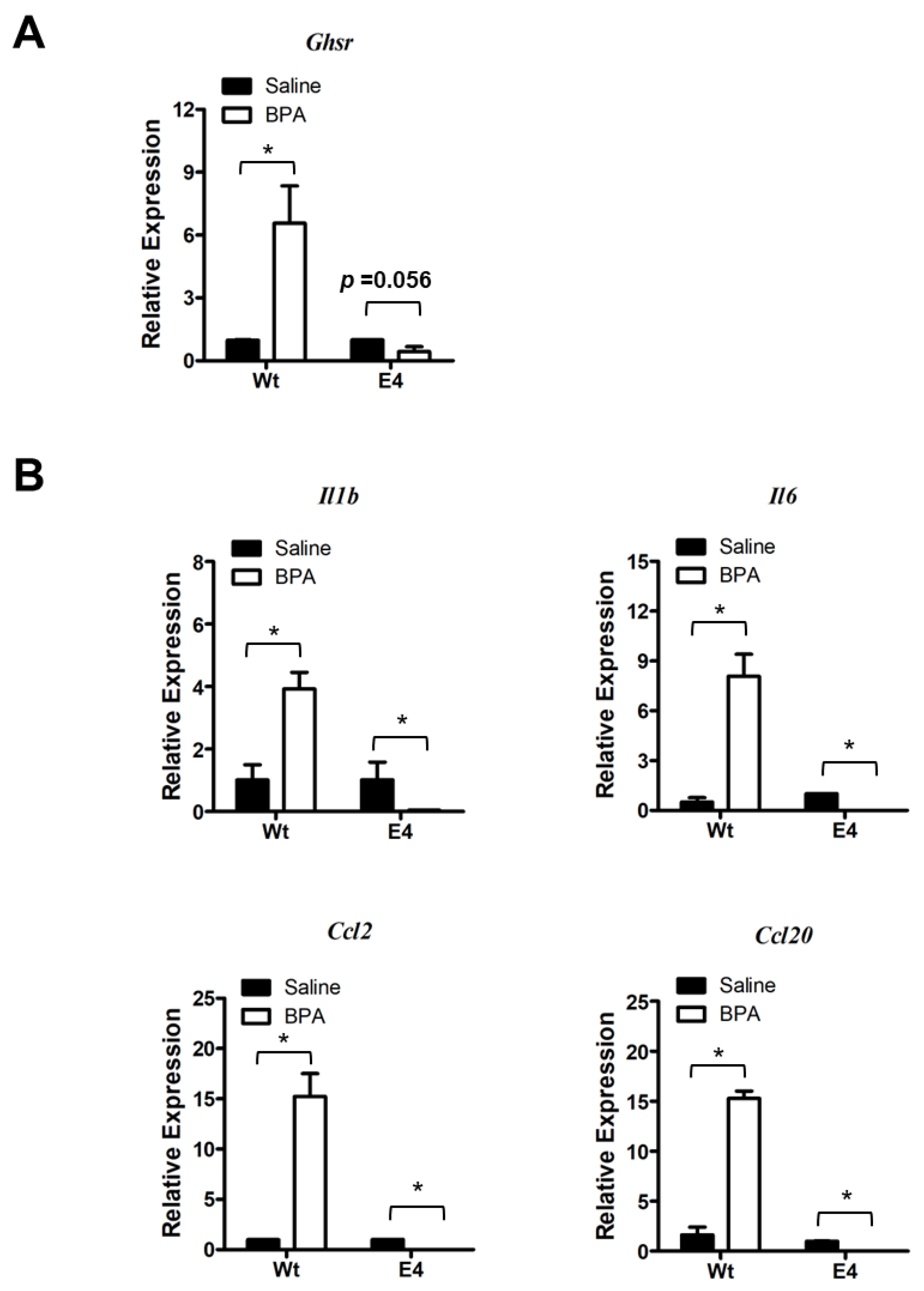
3.3. Ghsr Is Required for Pro-Inflammatory Activation of Macrophages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42, quiz e30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.H.; Achkar, J.P. Gene-environment interactions in inflammatory bowel disease pathogenesis. Curr. Opin. Gastroenterol. 2015, 31, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.M.; Lewis, J.D.; Mayer, E.A.; Plevy, S.E.; Chuang, E.; Rappaport, S.M.; Croitoru, K.; Korzenik, J.R.; Krischer, J.; Hyams, J.S.; et al. Challenges in IBD Research: Environmental Triggers. Inflamm. Bowel Dis. 2019, 25, S13–S23. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Khosravi, A.; Kusumawardhani, I.P.; Kwon, A.H.; Vasconcelos, A.C.; Cunha, L.D.; Mayer, A.E.; Shen, Y.; Wu, W.L.; Kambal, A.; et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016, 352, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Alomirah, H.; Cho, H.S.; Li, Y.F.; Liao, C.; Minh, T.B.; Mohd, M.A.; Nakata, H.; Ren, N.; Kannan, K. Urinary bisphenol A concentrations and their implications for human exposure in several Asian countries. Environ. Sci. Technol. 2011, 45, 7044–7050. [Google Scholar] [CrossRef]
- Welshons, W.V.; Nagel, S.C.; vom Saal, F.S. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology 2006, 147, S56–S69. [Google Scholar] [CrossRef] [Green Version]
- MacKay, H.; Abizaid, A. A plurality of molecular targets: The receptor ecosystem for bisphenol-A (BPA). Horm. Behav. 2018, 101, 59–67. [Google Scholar] [CrossRef]
- Linares, R.; Fernández, M.F.; Gutiérrez, A.; García-Villalba, R.; Suárez, B.; Zapater, P.; Martínez-Blázquez, J.A.; Caparrós, E.; Tomás-Barberán, F.A.; Francés, R. Endocrine disruption in Crohn’s disease: Bisphenol A enhances systemic inflammatory response in patients with gut barrier translocation of dysbiotic microbiota products. FASEB J. 2021, 35, e21697. [Google Scholar] [CrossRef]
- Chen, X.; Wang, S.; Mao, X.; Xiang, X.; Ye, S.; Chen, J.; Zhu, A.; Meng, Y.; Yang, X.; Peng, S.; et al. Adverse health effects of emerging contaminants on inflammatory bowel disease. Front. Public Health 2023, 11, 1140786. [Google Scholar] [CrossRef]
- Lang, I.A.; Galloway, T.S.; Scarlett, A.; Henley, W.E.; Depledge, M.; Wallace, R.B.; Melzer, D. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA 2008, 300, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Trasande, L.; Attina, T.M.; Blustein, J. Association between urinary bisphenol A concentration and obesity prevalence in children and adolescents. JAMA 2012, 308, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Chou, E.L.; Baecker, A.; You, N.C.; Song, Y.; Sun, Q.; Liu, S. Endocrine-disrupting chemicals, risk of type 2 diabetes, and diabetes-related metabolic traits: A systematic review and meta-analysis. J. Diabetes 2016, 8, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Morimoto, S.; Ripoll, C.; Fuentes, E.; Nadal, A. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ. Health Perspect. 2006, 114, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLuca, J.A.; Allred, K.F.; Menon, R.; Riordan, R.; Weeks, B.R.; Jayaraman, A.; Allred, C.D. Bisphenol-A alters microbiota metabolites derived from aromatic amino acids and worsens disease activity during colitis. Exp. Biol. Med. 2018, 243, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Malaise, Y.; Lencina, C.; Placide, F.; Bacquie, V.; Cartier, C.; Olier, M.; Buettner, M.; Wallbrecht, M.; Menard, S.; Guzylack-Piriou, L. Oral exposure to bisphenols induced food intolerance and colitis in vivo by modulating immune response in adult mice. Food Chem. Toxicol. 2020, 146, 111773. [Google Scholar] [CrossRef]
- Zhu, M.; Wei, R.; Li, Y.; Li, J.; Dong, M.; Chen, X.; Lv, L.; Qin, Z. Bisphenol chemicals disturb intestinal homeostasis via Notch/Wnt signaling and induce mucosal barrier dysregulation and inflammation. Sci. Total Environ. 2022, 828, 154444. [Google Scholar] [CrossRef]
- Yin, F.; Huang, X.; Lin, X.; Chan, T.F.; Lai, K.P.; Li, R. Analyzing the synergistic adverse effects of BPA and its substitute, BHPF, on ulcerative colitis through comparative metabolomics. Chemosphere 2022, 287, 132160. [Google Scholar] [CrossRef]
- Jones, G.R.; Bain, C.C.; Fenton, T.M.; Kelly, A.; Brown, S.L.; Ivens, A.C.; Travis, M.A.; Cook, P.C.; MacDonald, A.S. Dynamics of Colon Monocyte and Macrophage Activation During Colitis. Front. Immunol. 2018, 9, 2764. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, E.; Varol, C.; Farache, J.; Elmaliah, E.; Satpathy, A.T.; Friedlander, G.; Mack, M.; Shpigel, N.; Boneca, I.G.; Murphy, K.M.; et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity 2012, 37, 1076–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, N.; Hisamatsu, T.; Okamoto, S.; Chinen, H.; Kobayashi, T.; Sato, T.; Sakuraba, A.; Kitazume, M.T.; Sugita, A.; Koganei, K.; et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J. Clin. Invest. 2008, 118, 2269–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchanathan, R.; Liu, H.; Leung, Y.K.; Ho, S.M.; Choubey, D. Bisphenol A (BPA) stimulates the interferon signaling and activates the inflammasome activity in myeloid cells. Mol. Cell Endocrinol. 2015, 415, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Li, M.; Wu, C.; Zhou, C.; Zhang, J.; Zhu, Q.; Shen, T. Bisphenol A promotes macrophage proinflammatory subtype polarization via upregulation of IRF5 expression in vitro. Toxicol. In Vitro 2019, 60, 97–106. [Google Scholar] [CrossRef]
- Camarca, A.; Gianfrani, C.; Ariemma, F.; Cimmino, I.; Bruzzese, D.; Scerbo, R.; Picascia, S.; D’Esposito, V.; Beguinot, F.; Formisano, P.; et al. Human Peripheral Blood Mononuclear Cell Function and Dendritic Cell Differentiation Are Affected by Bisphenol-A Exposure. PLoS ONE 2016, 11, e0161122. [Google Scholar] [CrossRef] [Green Version]
- Damian, M.; Marie, J.; Leyris, J.P.; Fehrentz, J.A.; Verdie, P.; Martinez, J.; Baneres, J.L.; Mary, S. High constitutive activity is an intrinsic feature of ghrelin receptor protein: A study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J. Biol. Chem. 2012, 287, 3630–3641. [Google Scholar] [CrossRef] [Green Version]
- Mear, Y.; Enjalbert, A.; Thirion, S. GHS-R1a constitutive activity and its physiological relevance. Front. Neurosci. 2013, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Lee, J.H.; Buras, E.D.; Yu, K.; Wang, R.; Smith, C.W.; Wu, H.; Sheikh-Hamad, D.; Sun, Y. Ghrelin receptor regulates adipose tissue inflammation in aging. Aging 2016, 8, 178–191. [Google Scholar] [CrossRef] [Green Version]
- Waseem, T.; Duxbury, M.; Ito, H.; Ashley, S.W.; Robinson, M.K. Exogenous ghrelin modulates release of pro-inflammatory and anti-inflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Surgery 2008, 143, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Dixit, V.D.; Yang, H.; Sun, Y.; Weeraratna, A.T.; Youm, Y.H.; Smith, R.G.; Taub, D.D. Ghrelin promotes thymopoiesis during aging. J. Clin. Invest. 2007, 117, 2778–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, N.; Saito, T.; Yagyu, T.; Jiang, B.H.; Kitagawa, K.; Inagaki, C. GH, GH receptor, GH secretagogue receptor, and ghrelin expression in human T cells, B cells, and neutrophils. J. Clin. Endocrinol. Metab. 2001, 86, 4284–4291. [Google Scholar] [CrossRef]
- Noh, J.Y.; Herrera, M.; Patil, B.S.; Tan, X.D.; Wright, G.A.; Sun, Y. The expression and function of growth hormone secretagogue receptor in immune cells: A current perspective. Exp. Biol. Med. 2022, 247, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Goncalves, R.; Mosser, D.M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 2008, 83, 14. [Google Scholar] [CrossRef] [Green Version]
- Fuss, I.J.; Kanof, M.E.; Smith, P.D.; Zola, H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr. Protoc. Immunol. 2009, 85, 7. [Google Scholar] [CrossRef] [PubMed]
- Moolenbeek, C.; Ruitenberg, E.J. The “Swiss roll”: A simple technique for histological studies of the rodent intestine. Lab. Anim. 1981, 15, 57–59. [Google Scholar] [CrossRef]
- Erben, U.; Loddenkemper, C.; Doerfel, K.; Spieckermann, S.; Haller, D.; Heimesaat, M.M.; Zeitz, M.; Siegmund, B.; Kuhl, A.A. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 2014, 7, 4557–4576. [Google Scholar]
- Laustsen, A.; Bak, R.O. Electroporation-Based CRISPR/Cas9 Gene Editing Using Cas9 Protein and Chemically Modified sgRNAs. Methods Mol. Biol. 2019, 1961, 127–134. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency. Bisphenol A Action Plan (CASRN 80-05-7)—Bisphenol A (BPA) Summary. 2010. Available online: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/bisphenol-bpa-summary (accessed on 1 March 2023).
- Cassado Ados, A.; D’Imperio Lima, M.R.; Bortoluci, K.R. Revisiting mouse peritoneal macrophages: Heterogeneity, development, and function. Front. Immunol. 2015, 6, 225. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol. 2019, 20, 970–979. [Google Scholar] [CrossRef]
- Yamamoto-Furusho, J.K. Inflammatory bowel disease therapy: Blockade of cytokines and cytokine signaling pathways. Curr. Opin. Gastroenterol. 2018, 34, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Liu, P.; Phong, K.; Hinrichs, J.H.; Ataii, N.; Williams, K.; Hadler-Olsen, E.; Samson, S.; Gartner, Z.J.; Fisher, S.; et al. Bisphenol A replacement chemicals, BPF and BPS, induce protumorigenic changes in human mammary gland organoid morphology and proteome. Proc. Natl. Acad. Sci. USA 2022, 119, e2115308119. [Google Scholar] [CrossRef] [PubMed]
- Eladak, S.; Grisin, T.; Moison, D.; Guerquin, M.J.; N’Tumba-Byn, T.; Pozzi-Gaudin, S.; Benachi, A.; Livera, G.; Rouiller-Fabre, V.; Habert, R. A new chapter in the bisphenol A story: Bisphenol S and bisphenol F are not safe alternatives to this compound. Fertil. Steril. 2015, 103, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodila, A.; Franko, N.; Sollner Dolenc, M. A review on immunomodulatory effects of BPA analogues. Arch. Toxicol. 2023, 97, 1831–1846. [Google Scholar] [CrossRef] [PubMed]
- Ao, J.; Liu, Y.; Tang, W.; Zhang, J. Bisphenol S exposure induces intestinal inflammation: An integrated metabolomic and transcriptomic study. Chemosphere 2022, 292, 133510. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, W.; Ao, J.; Zhang, J.; Feng, L. Transcriptomics integrated with metabolomics reveals the effect of Bisphenol F (BPF) exposure on intestinal inflammation. Sci. Total. Environ. 2022, 816, 151644. [Google Scholar] [CrossRef]
- Gould, J.C.; Leonard, L.S.; Maness, S.C.; Wagner, B.L.; Conner, K.; Zacharewski, T.; Safe, S.; McDonnell, D.P.; Gaido, K.W. Bisphenol A interacts with the estrogen receptor alpha in a distinct manner from estradiol. Mol. Cell. Endocrinol. 1998, 142, 203–214. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Thomas, P.; Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 2006, 102, 175–179. [Google Scholar] [CrossRef]
- Jacenik, D.; Krajewska, W.M. Significance of G Protein-Coupled Estrogen Receptor in the Pathophysiology of Irritable Bowel Syndrome, Inflammatory Bowel Diseases and Colorectal Cancer. Front. Endocrinol. 2020, 11, 390. [Google Scholar] [CrossRef]
- Pelekanou, V.; Kampa, M.; Kiagiadaki, F.; Deli, A.; Theodoropoulos, P.; Agrogiannis, G.; Patsouris, E.; Tsapis, A.; Castanas, E.; Notas, G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERalpha36 and GPR30/GPER1. J. Leukoc. Biol. 2016, 99, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor CREB in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, R.P.; Guthrie, C.R.; Wailes, L.M.; Zhao, X.; Means, A.R.; McKnight, G.S. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol. Cell. Biol. 1994, 14, 6107–6116. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, X.; Liu, Z.; Han, H.W.; Noh, J.Y.; Shen, Z.; Kim, D.M.; Wang, H.; Guo, H.; Ballard, J.; Golovko, A.; et al. Nutrient-Sensing Ghrelin Receptor in Macrophages Modulates Bisphenol A-Induced Intestinal Inflammation in Mice. Genes 2023, 14, 1455. https://doi.org/10.3390/genes14071455
Ye X, Liu Z, Han HW, Noh JY, Shen Z, Kim DM, Wang H, Guo H, Ballard J, Golovko A, et al. Nutrient-Sensing Ghrelin Receptor in Macrophages Modulates Bisphenol A-Induced Intestinal Inflammation in Mice. Genes. 2023; 14(7):1455. https://doi.org/10.3390/genes14071455
Chicago/Turabian StyleYe, Xiangcang, Zeyu Liu, Hye Won Han, Ji Yeon Noh, Zheng Shen, Da Mi Kim, Hongying Wang, Huiping Guo, Johnathan Ballard, Andrei Golovko, and et al. 2023. "Nutrient-Sensing Ghrelin Receptor in Macrophages Modulates Bisphenol A-Induced Intestinal Inflammation in Mice" Genes 14, no. 7: 1455. https://doi.org/10.3390/genes14071455
APA StyleYe, X., Liu, Z., Han, H. W., Noh, J. Y., Shen, Z., Kim, D. M., Wang, H., Guo, H., Ballard, J., Golovko, A., Morpurgo, B., & Sun, Y. (2023). Nutrient-Sensing Ghrelin Receptor in Macrophages Modulates Bisphenol A-Induced Intestinal Inflammation in Mice. Genes, 14(7), 1455. https://doi.org/10.3390/genes14071455