
RALGAPA1 Deletion in Belgian Shepherd Dogs with Cerebellar Ataxia

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Examinations
2.2. Necropsy and Histology
2.3. Animals for Genetic Analysis
2.4. DNA Extraction
2.5. Linkage Analysis and Homozygosity Mapping
2.6. Whole-Genome Sequencing
2.7. Variant Calling and Filtering
2.8. PCR, Fragment Size Analysis and Sanger Sequencing
3. Results
3.1. Clinical Description
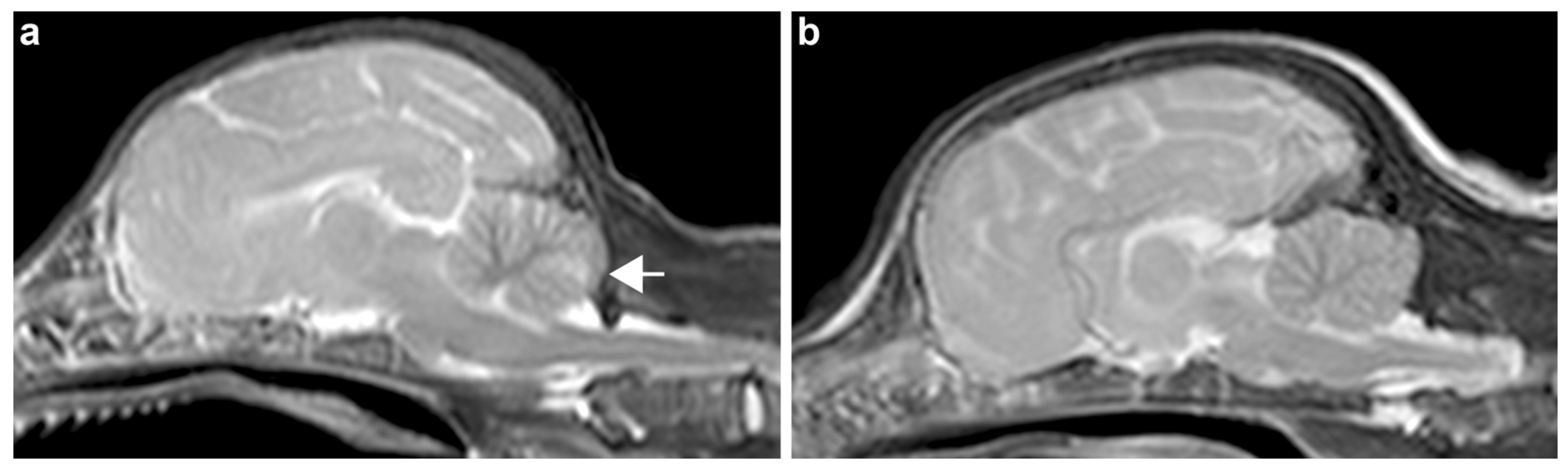
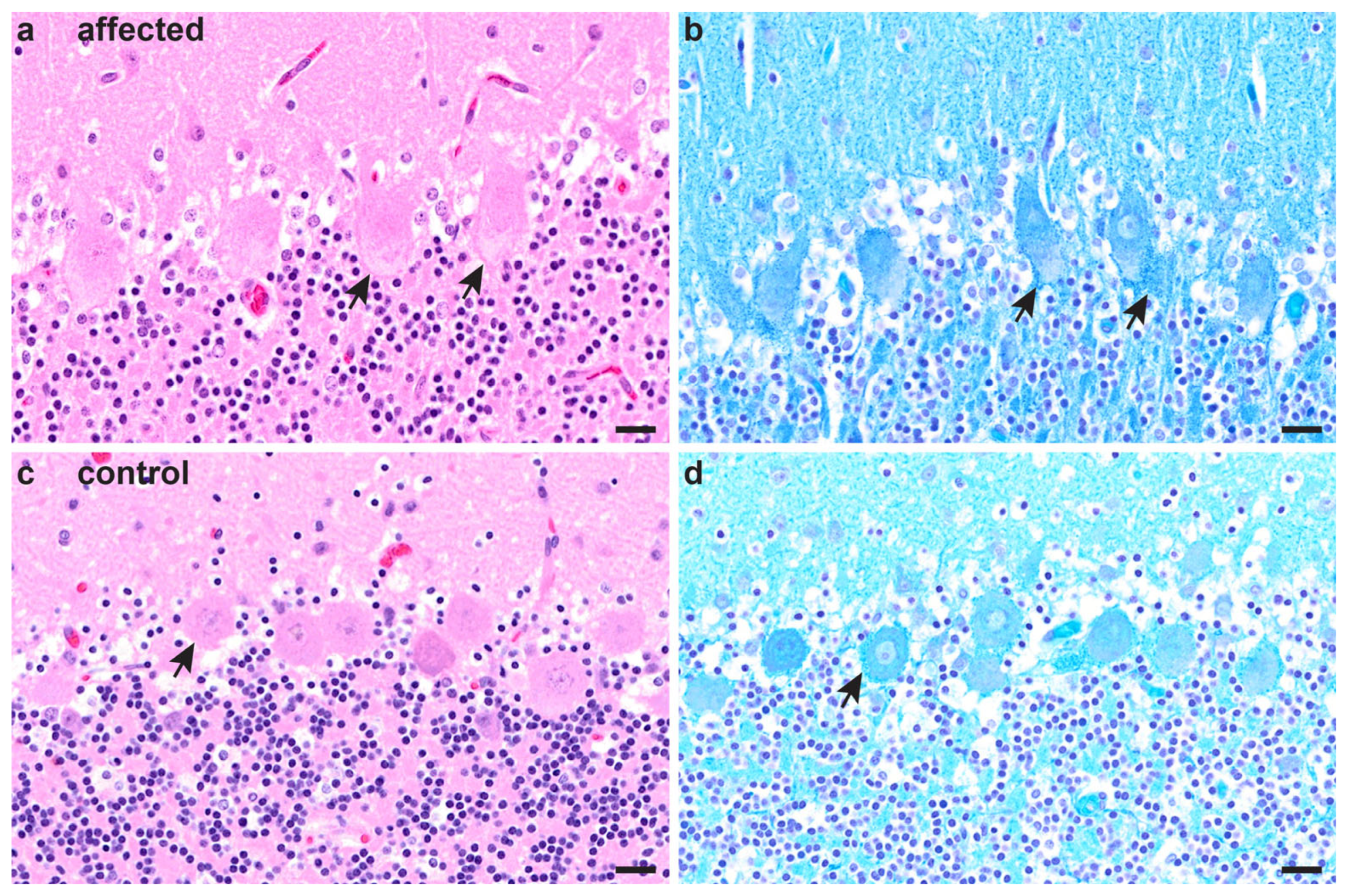
3.2. Necropsy and Histopathological Examinations
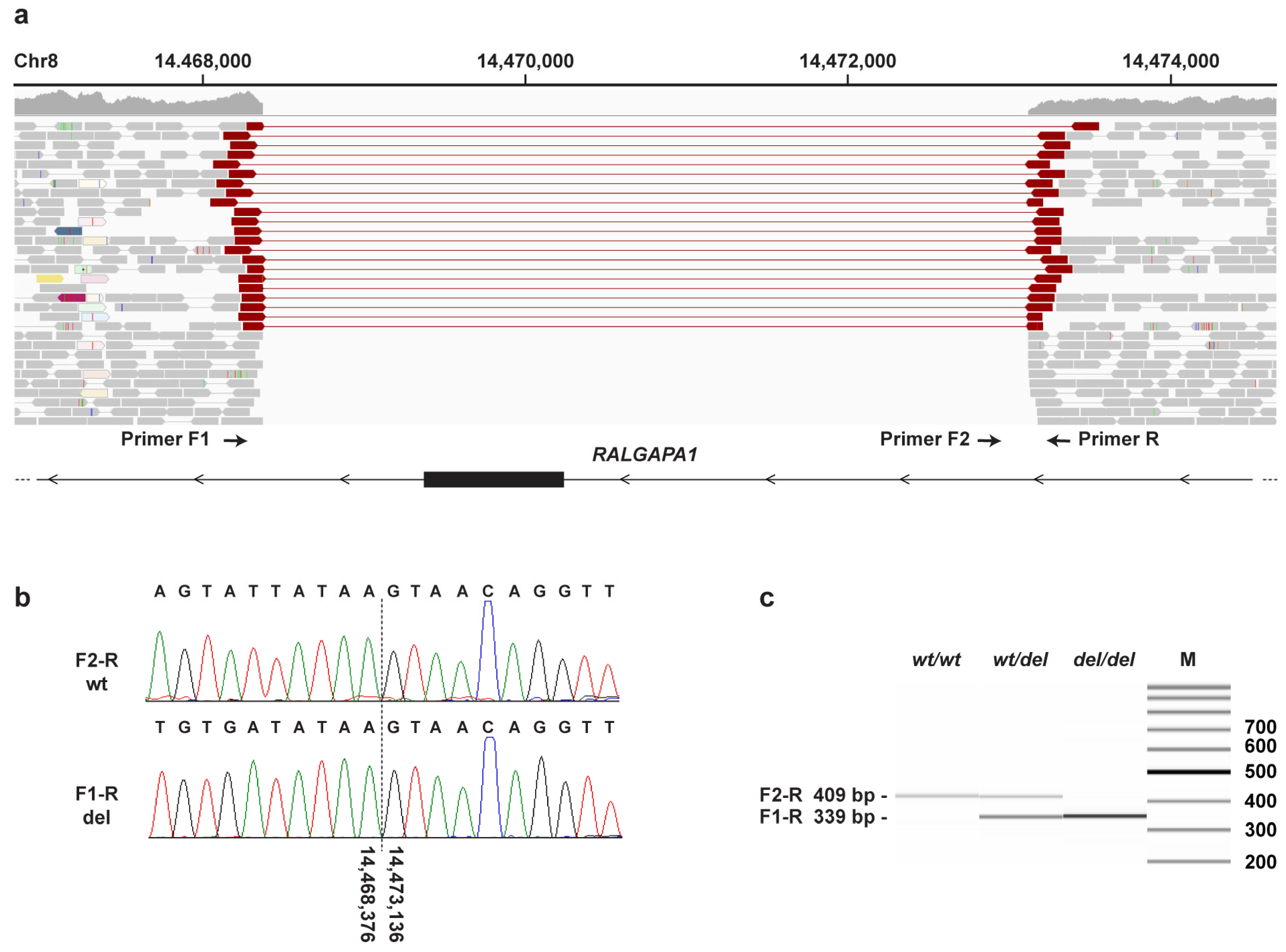
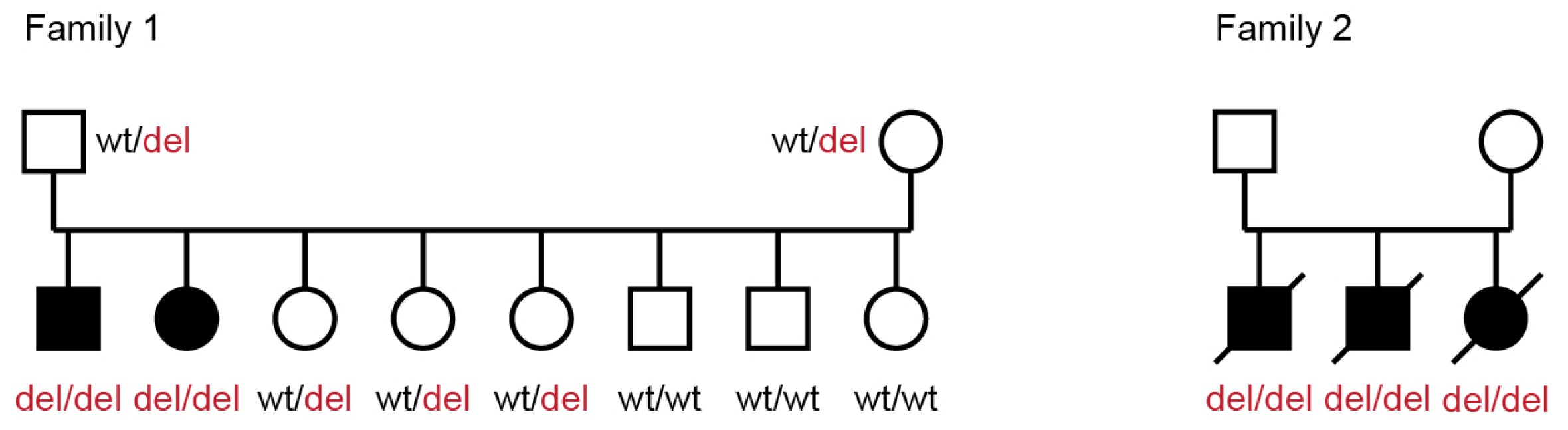
3.3. Genetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Urkasemsin, G.; Olby, N.J. Canine Hereditary Ataxia. Vet. Clin. N. Am. Small Anim. Pract. 2014, 44, 1075–1089. [Google Scholar] [CrossRef]
- Mellersh, C. Give a dog a genome. Vet. J. 2008, 178, 46–52. [Google Scholar] [CrossRef]
- Leroy, G. Genetic diversity, inbreeding and breeding practices in dogs: Results from pedigree analyses. Vet. J. 2011, 189, 177–182. [Google Scholar] [CrossRef]
- Cachin, M.; Vandevelde, M. Congenital Tremor with Spongy Degeneration of the Central Nervous System in Two Puppies. J. Vet. Intern. Med. 1991, 5, 87–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiter, M.; Högler, S.; Kneissl, S.; Url, A.; Leschnik, M. Spongy Degeneration with Cerebellar Ataxia in Malinois Puppies: A Hereditary Autosomal Recessive Disorder? J. Vet. Intern. Med. 2011, 25, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Mauri, N.; Kleiter, M.; Leschnik, M.; Högler, S.; Dietschi, E.; Wiedmer, M.; Dietrich, J.; Henke, D.; Steffen, F.; Schuller, S.; et al. A Missense Variant in KCNJ10 in Belgian Shepherd Dogs Affected by Spongy Degeneration with Cerebellar Ataxia (SDCA1). G3 Genes Genomes Genet. 2017, 7, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauri, N.; Kleiter, M.; Dietschi, E.; Leschnik, M.; Högler, S.; Wiedmer, M.; Dietrich, J.; Henke, D.; Steffen, F.; Schuller, S.; et al. A SINE Insertion in ATP1B2 in Belgian Shepherd Dogs Affected by Spongy Degeneration with Cerebellar Ataxia (SDCA2). G3 Genes Genomes Genet. 2017, 7, 2729–2737. [Google Scholar] [CrossRef] [Green Version]
- Van Poucke, M.; Stee, K.; Bhatti, S.F.M.; Vanhaesebrouck, A.; Bosseler, L.; Peelman, L.J.; Van Ham, L. The novel homozygous KCNJ10 c.986T>C (p.(Leu329Pro)) variant is pathogenic for the SeSAME/EAST homologue in Malinois dogs. Eur. J. Hum. Genet. 2017, 25, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Christen, M.; Högler, S.; Kleiter, M.; Leschnik, M.; Weber, C.; Thaller, D.; Jagannathan, V.; Leeb, T. Deletion of the SELENOP gene leads to CNS atrophy with cerebellar ataxia in dogs. PLoS Genet. 2021, 17, e1009716. [Google Scholar] [CrossRef]
- Van Poucke, M.; Stee, K.; Sonck, L.; Stock, E.; Bosseler, L.; Van Dorpe, J.; Van Nieuwerburgh, F.; Deforce, D.; Peelman, L.J.; Van Ham, L.; et al. Truncating SLC12A6 variants cause different clinical phenotypes in humans and dogs. Eur. J. Hum. Genet. 2019, 27, 1561–1568. [Google Scholar] [CrossRef]
- Žáková, P.; Makovický, P. Záchyt spinocerebelární ataxie u ATP1B2 heterozygotních Malinois štěňat: Kolik našich chovných Belgických ovčáků je přenašečů neurodegenerativních onemocnění ? Veterinarstvi 2021, 71, 17–23. [Google Scholar]
- Gregor, K.M.; Becker, S.C.; Hellhammer, F.; Schön, K.; Baumgärtner, W.; Puff, C. Histochemical staining techniques in Culex pipiens and Drosophila melanogaster (Diptera) with a comparison to mammals. Vet. Pathol. 2022, 59, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Aguirre, G.; André, C.; Bannasch, D.; Becker, D.; Davis, B.; Ekenstedt, K.; Faller, K.; et al. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakawa, R.; Fukai, S.; Kawato, M.; Higashi, T.; Kondo, H.; Ikeda, T.; Nakayama, E.; Okawa, K.; Nureki, O.; Kimura, T.; et al. Tuberous Sclerosis Tumor Suppressor Complex-like Complexes Act as GTPase-activating Proteins for Ral GTPases. J. Biol. Chem. 2009, 284, 21580–21588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.M.; Bodemann, B.O.; White, M.A. The RalGEF/Ral Pathway. Enzymes 2013, 34 Pt B, 137–156. [Google Scholar]
- Apken, L.H.; Oeckinghaus, A. The RAL signaling network: Cancer and beyond. In International Review of Cell and Molecular Biology; Elsevier Inc.: Amsterdam, The Netherlands, 2021; Volume 361, pp. 21–105. ISBN 9780128237571. [Google Scholar] [CrossRef]
- Richardson, D.S.; Spehar, J.M.; Han, D.T.; Chakravarthy, P.A.; Sizemore, S.T. The RAL Enigma: Distinct Roles of RALA and RALB in Cancer. Cells 2022, 11, 1645. [Google Scholar] [CrossRef]
- Peschard, P.; McCarthy, A.; Leblanc-Dominguez, V.; Yeo, M.; Guichard, S.; Stamp, G.; Marshall, C.J. Genetic deletion of RALA and RALB small GTPases reveals redundant functions in development and tumorigenesis. Curr. Biol. 2012, 22, 2063–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.; Skorobogatko, Y.; Pode-Shakked, B.; Powell, C.M.; Alhaddad, B.; Seibt, A.; Barel, O.; Heimer, G.; Hoffmann, C.; Demmer, L.A.; et al. Bi-allelic Variants in RALGAPA1 Cause Profound Neurodevelopmental Disability, Muscular Hypotonia, Infantile Spasms, and Feeding Abnormalities. Am. J. Hum. Genet. 2020, 106, 246–255. [Google Scholar] [CrossRef]
- Shimojima, K.; Komoike, Y.; Tohyama, J.; Takahashi, S.; Páez, M.T.; Nakagawa, E.; Goto, Y.; Ohno, K.; Ohtsu, M.; Oguni, H.; et al. TULIP1 (RALGAPA1) haploinsufficiency with brain development delay. Genomics 2009, 94, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Phemister, R.D.; Young, S. The postnatal development of the canine cerebellar cortex. J. Comp. Neurol. 1968, 134, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype (894 Dogs) | wt/wt | wt/del | del/del |
|---|---|---|---|
| Cerebellar ataxia cases from family 1 (n = 2) | - | - | 2 |
| Unaffected members of family 1 (n = 8) | 3 | 5 | - |
| SDCA1, SDCA2, CACA cases (n = 21) | 21 | - | - |
| Unexplained archived ataxia cases (n = 36) | 32 | 1 | 3 1 |
| Unrelated controls (n = 827) | 785 | 42 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christen, M.; Zdora, I.; Leschnik, M.; Jagannathan, V.; Puff, C.; Hünerfauth, E.; Volk, H.A.; Baumgärtner, W.; Koch, T.C.; Schäfer, W.; et al. RALGAPA1 Deletion in Belgian Shepherd Dogs with Cerebellar Ataxia. Genes 2023, 14, 1520. https://doi.org/10.3390/genes14081520
Christen M, Zdora I, Leschnik M, Jagannathan V, Puff C, Hünerfauth E, Volk HA, Baumgärtner W, Koch TC, Schäfer W, et al. RALGAPA1 Deletion in Belgian Shepherd Dogs with Cerebellar Ataxia. Genes. 2023; 14(8):1520. https://doi.org/10.3390/genes14081520
Chicago/Turabian StyleChristen, Matthias, Isabel Zdora, Michael Leschnik, Vidhya Jagannathan, Christina Puff, Enrice Hünerfauth, Holger A. Volk, Wolfgang Baumgärtner, Tessa C. Koch, Wencke Schäfer, and et al. 2023. "RALGAPA1 Deletion in Belgian Shepherd Dogs with Cerebellar Ataxia" Genes 14, no. 8: 1520. https://doi.org/10.3390/genes14081520
APA StyleChristen, M., Zdora, I., Leschnik, M., Jagannathan, V., Puff, C., Hünerfauth, E., Volk, H. A., Baumgärtner, W., Koch, T. C., Schäfer, W., Kleiter, M., & Leeb, T. (2023). RALGAPA1 Deletion in Belgian Shepherd Dogs with Cerebellar Ataxia. Genes, 14(8), 1520. https://doi.org/10.3390/genes14081520