
Impact of High-to-Moderate Penetrance Genes on Genetic Testing: Looking over Breast Cancer

 , , , , ,
, , , , ,  , add
Show full author list
, add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients’ Recruitment
2.2. Next-Generation Sequencing (NGS)
2.3. Genetic Variant Classification
2.4. Sanger Sequencing
2.5. Copy Number Variants (CNVs) Detection
3. Results
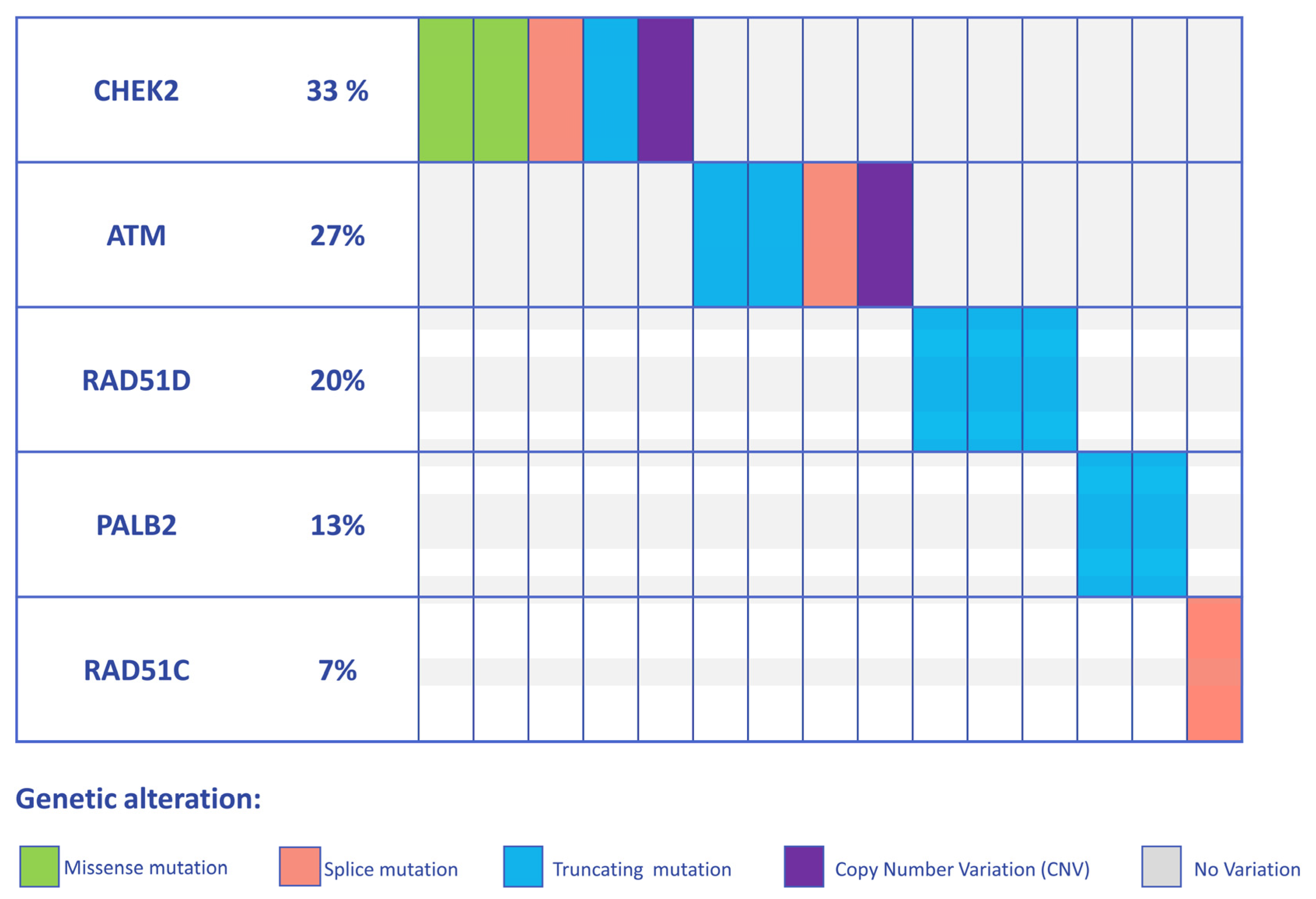
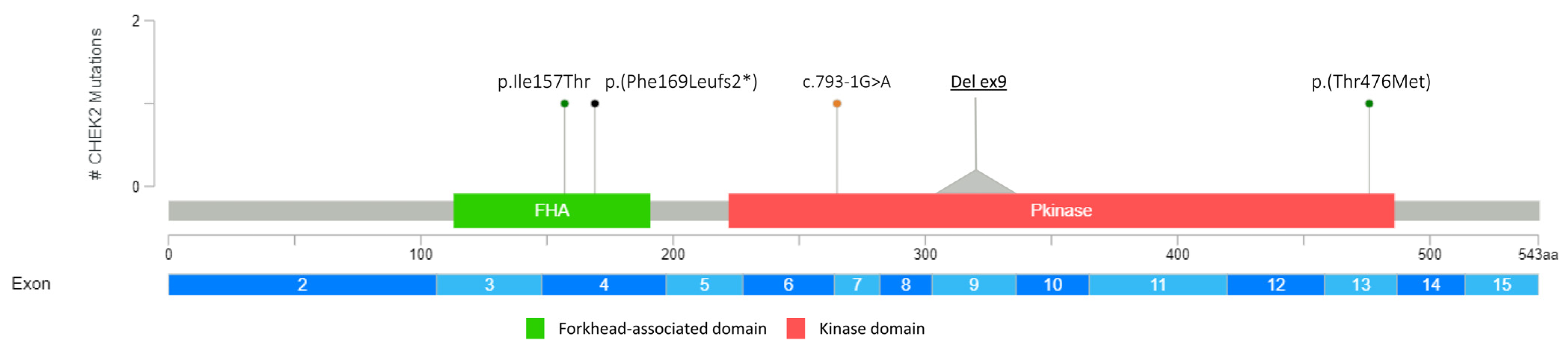
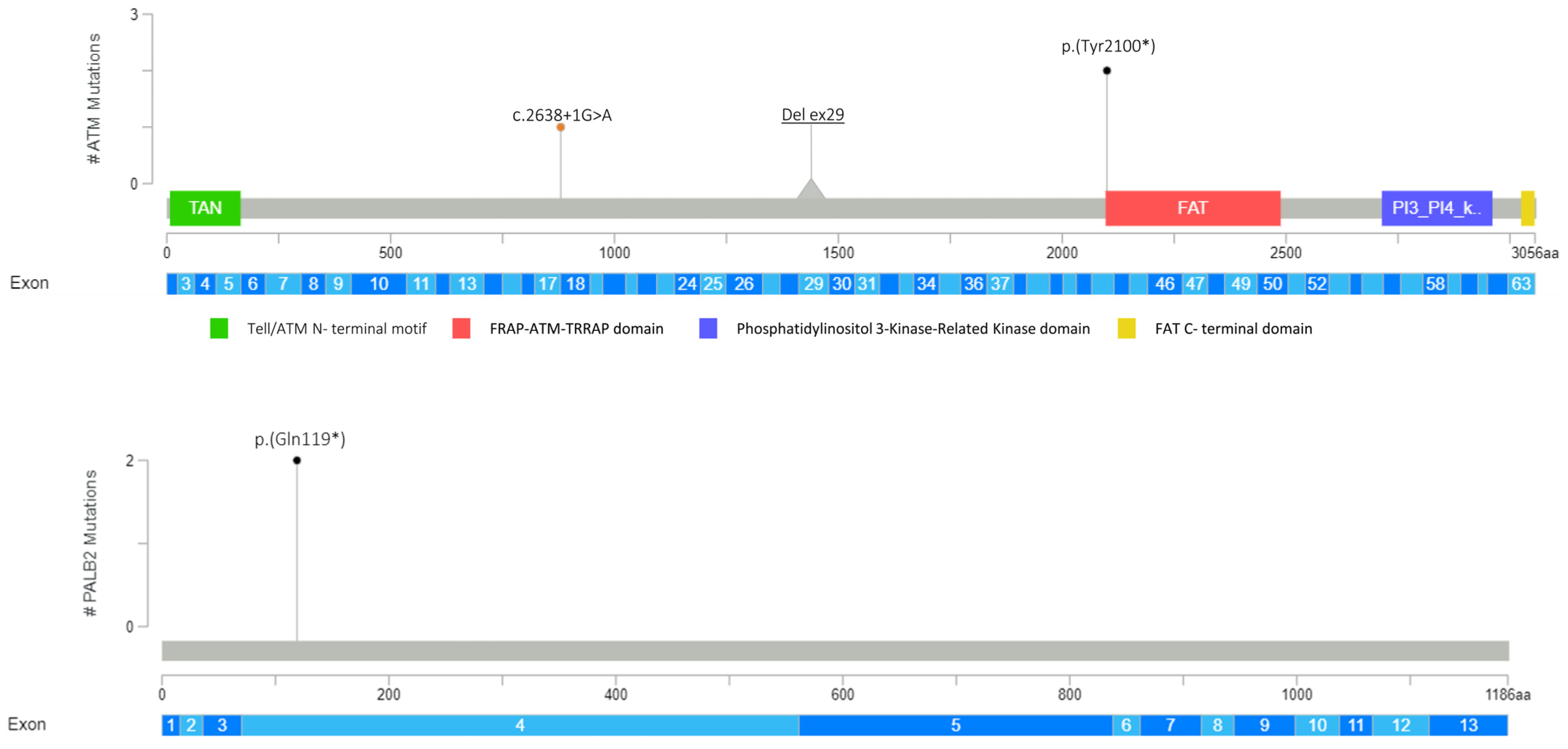
3.1. Detection of PVs and LPVs in BC Moderate Penetrance Genes
3.2. Clinical and Demographic Characteristics of High-to-Moderate Penetrance Gene Positive Patients
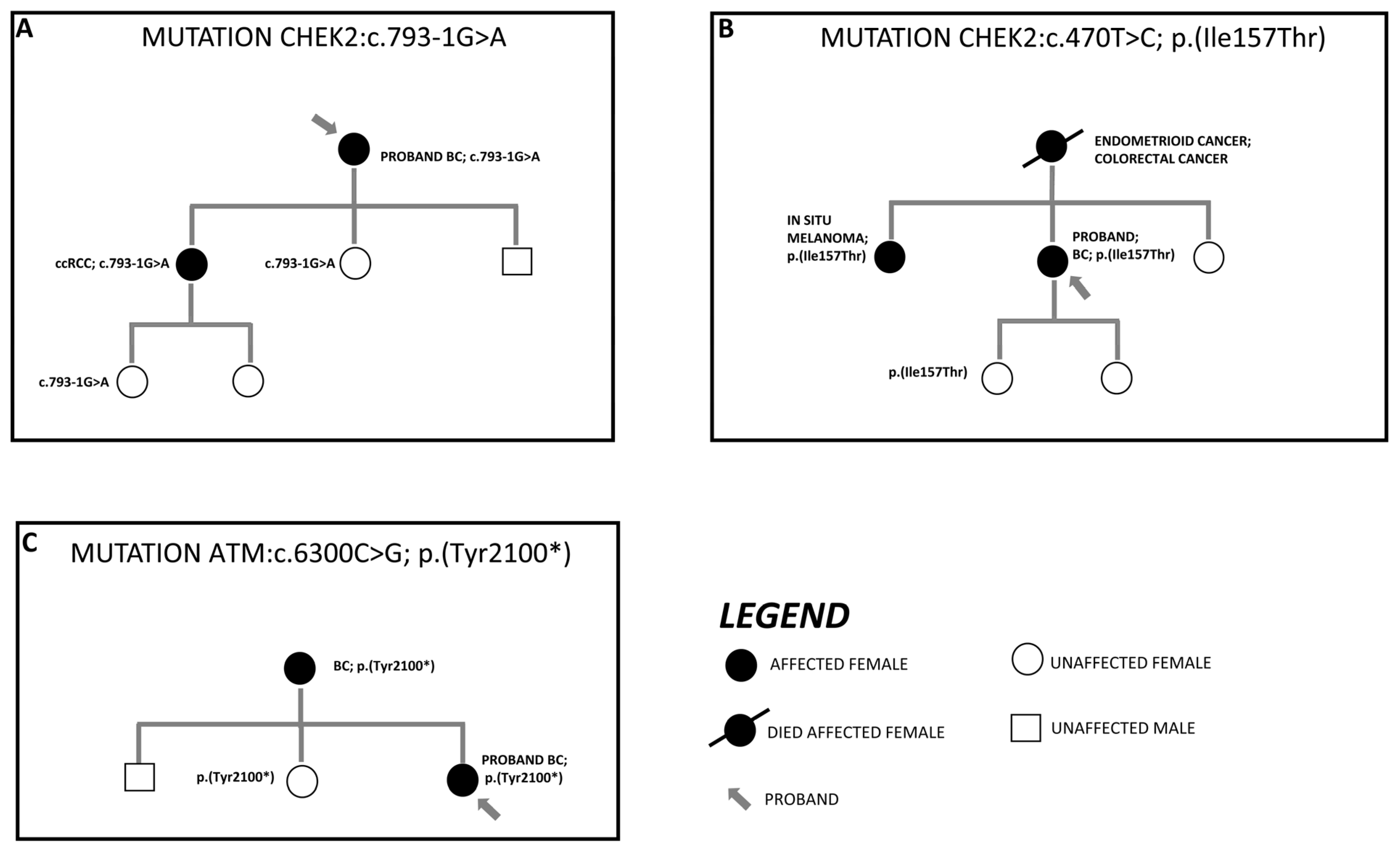
3.3. Segregation Analysis of Detected Variant in First-Degree Family Members
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hutton, M.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 77–102. [Google Scholar]
- Fanale, D.; Incorvaia, L.; Filorizzo, C.; Bono, M.; Fiorino, A.; Calò, V.; Brando, C.; Corsini, L.R.; Barraco, N.; Badalamenti, G.; et al. Detection of Germline Mutations in a Cohort of 139 Patients with Bilateral Breast Cancer by Multi-Gene Panel Testing: Impact of Pathogenic Variants in Other Genes beyond BRCA1/2. Cancers 2020, 12, 2415. [Google Scholar] [PubMed]
- Shah, P.D.; Patil, S.; Dickler, M.N.; Offit, K.; Hudis, C.A.; Robson, M.E. Twenty-One-Gene Recurrence Score Assay in BRCA-Associated versus Sporadic Breast Cancers: Differences Based on Germline Mutation Status. Cancer 2016, 122, 1178–1184. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Fountzilas, C.; Kaklamani, V.G. Multi-Gene Panel Testing in Breast Cancer Management. In Optimizing Breast Cancer Management; Cancer Treatment and Research Series; Springer: Cham, Switzerland, 2018; Volume 173, pp. 121–140. [Google Scholar]
- Neben, C.L.; Zimmer, A.D.; Stedden, W.; van den Akker, J.; O’Connor, R.; Chan, R.C.; Chen, E.; Tan, Z.; Leon, A.; Ji, J.; et al. Multi-Gene Panel Testing of 23,179 Individuals for Hereditary Cancer Risk Identifies Pathogenic Variant Carriers Missed by Current Genetic Testing Guidelines. J. Mol. Diagn. 2019, 21, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; Mcgowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Tenedini, E.; Piombino, C.; Venturelli, M.; Marchi, I.; Gasparini, E.; Barbieri, E.; Razzaboni, E.; Domati, F.; Caggia, F.; et al. Clinicopathologic Profile of Breast Cancer in Germline ATM and CHEK2 Mutation Carriers. Genes 2021, 12, 616. [Google Scholar] [CrossRef]
- Hauke, J.; Horvath, J.; Groß, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene Panel Testing of 5589 BRCA1/2-Negative Index Patients with Breast Cancer in a Routine Diagnostic Setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358. [Google Scholar] [CrossRef]
- Weitzel, J.N.; Neuhausen, S.L.; Adamson, A.; Tao, S.; Ricker, C.; Maoz, A.; Rosenblatt, M.; Nehoray, B.; Sand, S.; Steele, L.; et al. Pathogenic and Likely Pathogenic Variants in PALB2, CHEK2, and Other Known Breast Cancer Susceptibility Genes among 1054 BRCA-Negative Hispanics with Breast Cancer. Cancer 2019, 125, 2829–2836. [Google Scholar] [CrossRef]
- Park, J.S.; Shin, S.; Lee, Y.J.; Lee, S.T.; Nam, E.J.; Han, J.W.; Lee, S.H.; Kim, T.I.; Park, H.S. Implication and Influence of Multigene Panel Testing with Genetic Counseling in Korean Patients with BRCA1/2 Mutation-Negative Breast Cancer. Cancer Res. Treat. 2022, 54, 1099–1110. [Google Scholar] [CrossRef]
- Bono, M.; Fanale, D.; Incorvaia, L.; Cancelliere, D.; Fiorino, A.; Calò, V.; Dimino, A.; Filorizzo, C.; Corsini, L.R.; Brando, C.; et al. Impact of Deleterious Variants in Other Genes beyond BRCA1/2 Detected in Breast/Ovarian and Pancreatic Cancer Patients by NGS-Based Multi-Gene Panel Testing: Looking over the Hedge. ESMO Open 2021, 6, 100235. [Google Scholar] [CrossRef]
- Kraus, C.; Hoyer, J.; Vasileiou, G.; Wunderle, M.; Lux, M.P.; Fasching, P.A.; Krumbiegel, M.; Uebe, S.; Reuter, M.; Beckmann, M.W.; et al. Gene Panel Sequencing in Familial Breast/Ovarian Cancer Patients Identifies Multiple Novel Mutations Also in Genes Others than BRCA1/2. Int. J. Cancer 2017, 140, 95–102. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline Mutations in RAD51D Confer Susceptibility to Ovarian Cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.L.; Schwab, R.B.; Wallace, A.M.; Madlensky, L. Breast Cancer in a RAD51D Mutation Carrier: Case Report and Review of the Literature. Clin. Breast Cancer 2015, 15, e71–e75. [Google Scholar] [CrossRef]
- Thompson, E.R.; Rowley, S.M.; Sawyer, S.; Eccles, D.M.; Trainer, A.H.; Mitchell, G.; James, P.A.; Campbell, I.G. Analysis of RAD51D in Ovarian Cancer Patients and Families with a History of Ovarian or Breast Cancer. PLoS ONE 2013, 8, e54772. [Google Scholar] [CrossRef]
- Akcay, I.M.; Celik, E.; Agaoglu, N.B.; Alkurt, G.; Kizilboga Akgun, T.; Yildiz, J.; Enc, F.; Kir, G.; Canbek, S.; Kilic, A.; et al. Germline Pathogenic Variant Spectrum in 25 Cancer Susceptibility Genes in Turkish Breast and Colorectal Cancer Patients and Elderly Controls. Int. J. Cancer 2021, 148, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Muranen, T.A.; Blomqvist, C.; Dörk, T.; Jakubowska, A.; Heikkilä, P.; Fagerholm, R.; Greco, D.; Aittomäki, K.; Bojesen, S.E.; Shah, M.; et al. Patient Survival and Tumor Characteristics Associated with CHEK2:P.I157T—Findings from the Breast Cancer Association Consortium. Breast Cancer Res. 2016, 18, 98. [Google Scholar] [CrossRef] [PubMed]
- Bychkovsky, B.L.; Agaoglu, N.B.; Horton, C.; Zhou, J.; Yussuf, A.; Hemyari, P.; Richardson, M.E.; Young, C.; Laduca, H.; McGuinness, D.L.; et al. Differences in Cancer Phenotypes among Frequent CHEK2 Variants and Implications for Clinical Care—Checking CHEK2. JAMA Oncol. 2022, 8, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Mundt, E.; Nix, P.; Brown, K.; Bowles, K.R.; Manley, S. Complexities of Variant Classification in Clinical Hereditary Cancer Genetic Testing. J. Clin. Oncol. 2017, 35, 3796–3799. [Google Scholar] [CrossRef] [PubMed]
- Boonen, R.A.C.M.; Wiegant, W.W.; Celosse, N.; Vroling, B.; Heijl, S.; Kote-Jara, Z.; Mijuskovic, M.; Cristea, S.; Solleveld-Westerin, N.; Van Wezel, T.; et al. Functional Analysis Identifies Damaging CHEK2 Missense Variants Associated with Increased Cancer Risk. Cancer Res. 2022, 82, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Kleiblova, P.; Stolarova, L.; Krizova, K.; Lhota, F.; Hojny, J.; Zemankova, P.; Havranek, O.; Vocka, M.; Cerna, M.; Lhotova, K.; et al. Identification of Deleterious Germline CHEK2 Mutations and Their Association with Breast and Ovarian Cancer. Int. J. Cancer 2019, 145, 1782–1797. [Google Scholar] [CrossRef]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A Checkpoint Pathway Guards against Radioresistant DNA Synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef]
- Falck, J.; Lukas, C.; Protopopova, M.; Lukas, J.; Selivanova, G.; Bartek, J. Functional Impact of Concomitant versus Alternative Defects in the Chk2-P53 Tumour Suppressor Pathway. Oncogene 2001, 20, 5503–5510. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.Y.; Li, X.; Davis, H.L.; Canman, C.E. Phosphorylation of Threonine 68 Promotes Oligomerization and Autophosphorylation of the Chk2 Protein Kinase via the Forkhead-Associated Domain. J. Biol. Chem. 2002, 277, 19389–19395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpivaara, O.; Vahteristo, P.; Falck, J.; Syrjäkoski, K.; Eerola, H.; Easton, D.; Bartkova, J.; Lukas, J.; Heikkilä, P.; Aittomäki, K.; et al. CHEK2 Variant I157T May Be Associated with Increased Breast Cancer Risk. Int. J. Cancer 2004, 111, 543–547. [Google Scholar] [CrossRef]
- Schwarz, J.K.; Lovly, C.M.; Piwnica-Worms, H. Regulation of the Chk2 Protein Kinase by Oligomerization-Mediated Cis- and Trans-Phosphorylation. Mol. Cancer Res. 2003, 1, 598–609. [Google Scholar]
- Cai, Z.; Chehab, N.H.; Pavletich, N.P. Structure and Activation Mechanism of the CHK2 DNA Damage Checkpoint Kinase. Mol. Cell 2009, 35, 818–829. [Google Scholar] [CrossRef]
- Agaoglu, N.B.; Unal, B.; Akgun Dogan, O.; Kanev, M.O.; Zolfagharian, P.; Ozemri Sag, S.; Temel, S.G.; Doganay, L. Consistency of Variant Interpretations among Bioinformaticians and Clinical Geneticists in Hereditary Cancer Panels. Eur. J. Hum. Genet. 2022, 30, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Desrichard, A.; Bidet, Y.; Uhrhammer, N.; Bignon, Y.J. CHEK2 Contribution to Hereditary Breast Cancer in Non-BRCA Families. Breast Cancer Res. 2011, 13, R119. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Wokołorczyk, D.; Huzarski, T.; Byrski, T.; Gronwald, J.; Górski, B.; Dębniak, T.; Masojć, B.; Jakubowska, A.; Van De Wetering, T.; et al. A Deletion in CHEK2 of 5,395 Bp Predisposes to Breast Cancer in Poland. Breast Cancer Res. Treat. 2007, 102, 119–122. [Google Scholar] [CrossRef]
- Nizic-Kos, T.; Krajc, M.; Blatnik, A.; Stegel, V.; Skerl, P.; Novakovic, S.; Gazic, B.; Besic, N. Bilateral Disease Common among Slovenian CHEK2-Positive Breast Cancer Patients. Ann. Surg. Oncol. 2021, 28, 2561–2570. [Google Scholar] [CrossRef]
- Dennis, J.; Tyrer, J.P.; Walker, L.C.; Michailidou, K.; Dorling, L.; Bolla, M.K.; Wang, Q.; Ahearn, T.U.; Andrulis, I.L.; Anton-Culver, H.; et al. Rare Germline Copy Number Variants (CNVs) and Breast Cancer Risk. Commun. Biol. 2022, 5, 65. [Google Scholar] [CrossRef]
- Bandeira, G.; Rocha, K.; Lazar, M.; Ezquina, S.; Yamamoto, G.; Varela, M.; Takahashi, V.; Aguena, M.; Gollop, T.; Zatz, M.; et al. Germline Variants of Brazilian Women with Breast Cancer and Detection of a Novel Pathogenic ATM Deletion in Early-Onset Breast Cancer. Breast Cancer 2021, 28, 346–354. [Google Scholar] [CrossRef]
- Parenti, S.; Rabacchi, C.; Marino, M.; Tenedini, E.; Artuso, L.; Castellano, S.; Carretta, C.; Mallia, S.; Cortesi, L.; Toss, A.; et al. Characterization of New ATM Deletion Associated with Hereditary Breast Cancer. Genes 2021, 12, 136. [Google Scholar] [CrossRef]
- Golmard, L.; Castéra, L.; Krieger, S.; Moncoutier, V.; Abidallah, K.; Tenreiro, H.; Laugé, A.; Tarabeux, J.; Millot, G.A.; Nicolas, A.; et al. Contribution of Germline Deleterious Variants in the RAD51 Paralogs to Breast and Ovarian Cancers /631/208/68 /631/67/1347 Article. Eur. J. Hum. Genet. 2017, 25, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Vuorela, M.; Pylkäs, K.; Hartikainen, J.M.; Sundfeldt, K.; Lindblom, A.; Von Wachenfeldt Wäppling, A.; Haanpää, M.; Puistola, U.; Rosengren, A.; Anttila, M.; et al. Further Evidence for the Contribution of the RAD51C Gene in Hereditary Breast and Ovarian Cancer Susceptibility. Breast Cancer Res. Treat. 2011, 130, 1003–1010. [Google Scholar] [CrossRef]
- Schnurbein, G.; Hauke, J.; Wappenschmidt, B.; Weber-Lassalle, N.; Engert, S.; Hellebrand, H.; Garbes, L.; Becker, A.; Neidhardt, G.; Rhiem, K.; et al. RAD51C Deletion Screening Identifies a Recurrent Gross Deletion in Breast Cancer and Ovarian Cancer Families. Breast Cancer Res. 2013, 15, R120. [Google Scholar]
- Yang, X.; Song, H.; Leslie, G.; Engel, C.; Hahnen, E.; Auber, B.; Horváth, J.; Kast, K.; Niederacher, D.; Turnbull, C.; et al. Ovarian and Breast Cancer Risks Associated with Pathogenic Variants in RAD51C and RAD51D. J. Natl. Cancer Inst. 2020, 112, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef] [Green Version]
- Tsai, G.J.; Rañola, J.M.O.; Smith, C.; Garrett, L.T.; Bergquist, T.; Casadei, S.; Bowen, D.J.; Shirts, B.H. Outcomes of 92 Patient-Driven Family Studies for Reclassification of Variants of Uncertain Significance. Genet. Med. 2019, 21, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Golmard, L.; Caux-Moncoutier, V.; Davy, G.; Al Ageeli, E.; Poirot, B.; Tirapo, C.; Michaux, D.; Barbaroux, C.; d’Enghien, C.D.; Nicolas, A.; et al. Germline Mutation in the RAD51B Gene Confers Predisposition to Breast Cancer. BMC Cancer 2013, 13, 484. [Google Scholar] [CrossRef]
- Casadei, S.; Gulsuner, S.; Shirts, B.H.; Mandell, J.B.; Kortbawi, H.M.; Norquist, B.S.; Swisher, E.M.; Lee, M.K.; Goldberg, Y.; O’Connor, R.; et al. Characterization of Splice-Altering Mutations in Inherited Predisposition to Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 26798–26807. [Google Scholar] [CrossRef]
- Balmaña, J.; Digiovanni, L.; Gaddam, P.; Walsh, M.F.; Joseph, V.; Stadler, Z.K.; Nathanson, K.L.; Garber, J.E.; Couch, F.J.; Offit, K.; et al. Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. J. Clin. Oncol. 2016, 34, 4071–4078. [Google Scholar] [CrossRef] [PubMed]
- Jarhelle, E.; Riise Stensland, H.M.F.; Hansen, G.Å.M.; Skarsfjord, S.; Jonsrud, C.; Ingebrigtsen, M.; Strømsvik, N.; Van Ghelue, M. Identifying Sequence Variants Contributing to Hereditary Breast and Ovarian Cancer in BRCA1 and BRCA2 Negative Breast and Ovarian Cancer Patients. Sci. Rep. 2019, 9, 19986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanoguera-Miralles, L.; Valenzuela-Palomo, A.; Bueno-Martínez, E.; Llovet, P.; Díez-Gómez, B.; Caloca, M.J.; Pérez-Segura, P.; Fraile-Bethencourt, E.; Colmena, M.; Carvalho, S.; et al. Comprehensive Functional Characterization and Clinical Interpretation of 20 Splice-Site Variants of the Rad51c Gene. Cancers 2020, 12, 3771. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.; Van Asperen, C.J.; Jacobi, C.E.; Vink, G.R.; Tibben, A.; Breuning, M.H.; Otten, W. Variants of Uncertain Clinical Significance as a Result of BRCA1/2 Testing: Impact of an Ambiguous Breast Cancer Risk Message. Genet. Test. 2004, 8, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Culver, J.; Brinkerhoff, C.; Clague, J.; Yang, K.; Singh, K.; Sand, S.; Weitzel, J. Variants of Uncertain Significance in BRCA Testing: Evaluation of Surgical Decisions, Risk Perception, and Cancer Distress. Clin. Genet. 2013, 84, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, A.N.; LeBoeuf, N.R.; Nambudiri, V.E. Skin Cancer Risk in CHEK2 Mutation Carriers. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 353–359. [Google Scholar] [CrossRef]
- Smith, P.S.; West, H.; Whitworth, J.; Castle, B.; Sansbury, F.H.; Warren, A.Y.; Woodward, E.R.; Tischkowitz, M.; Maher, E.R. Pathogenic Germline Variants in Patients with Features of Hereditary Renal Cell Carcinoma: Evidence for Further Locus Heterogeneity. Genes Chromosomes Cancer 2021, 60, 5–16. [Google Scholar] [CrossRef]
- Hartman, T.R.; Demidova, E.V.; Lesh, R.W.; Hoang, L.; Richardson, M.; Forman, A.; Kessler, L.; Speare, V.; Golemis, E.A.; Hall, M.J.; et al. Prevalence of Pathogenic Variants in DNA Damage Response and Repair Genes in Patients Undergoing Cancer Risk Assessment and Reporting a Personal History of Early-Onset Renal Cancer. Sci. Rep. 2020, 10, 13518. [Google Scholar] [CrossRef]
- Ged, Y.; Chaim, J.L.; DInatale, R.G.; Knezevic, A.; Kotecha, R.R.; Carlo, M.I.; Lee, C.H.; Foster, A.; Feldman, D.R.; Teo, M.Y.; et al. DNA Damage Repair Pathway Alterations in Metastatic Clear Cell Renal Cell Carcinoma and Implications on Systemic Therapy. J. Immunother. Cancer 2020, 8, e000230. [Google Scholar] [CrossRef]
- Carlo, M.I.; Mukherjee, S.; Mandelker, D.; Vijai, J.; Kemel, Y.; Zhang, L.; Knezevic, A.; Patil, S.; Ceyhan-Birsoy, O.; Huang, K.C.; et al. Prevalence of Germline Mutations in Cancer Susceptibility Genes in Patients with Advanced Renal Cell Carcinoma. JAMA Oncol. 2018, 4, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef]
- Cybulski, C.; Górski, B.; Huzarski, T.; Masojć, B.; Mierzejewski, M.; Dȩbniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 Is a Multiorgan Cancer Susceptibility Gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Brooks, K.; Holman, M.; Steding, C.; Tucker, M. A Founder CHEK2 Pathogenic Variant in Association with Kidney Cancer. Cancer Genet. 2022, 262–263, 40–42. [Google Scholar] [CrossRef]
- Bergstrom, C.; Pence, C.; Berg, J.; Partain, N.; Sadeghi, N.; Mauer, C.; Pirzadeh-Miller, S.; Gao, A.; Li, H.; Unni, N.; et al. Clinicopathological Features and Outcomes in Individuals with Breast Cancer and ATM, CHEK2, or PALB2 Mutations. Ann. Surg. Oncol. 2021, 28, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xie, Y. Associations between RAD51D Germline Mutations and Breast Cancer Risk and Survival in BRCA1/2-Negative Breast Cancers. Ann. Oncol. 2018, 29, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Alizart, M.; Saunus, J.; Cummings, M.; Lakhani, S.R. Molecular Classification of Breast Carcinoma. Diagn. Histopathol. 2012, 18, 97–103. [Google Scholar] [CrossRef]
- Sokolova, A.; Johnstone, K.J.; McCart Reed, A.E.; Simpson, P.T.; Lakhani, S.R. Hereditary Breast Cancer: Syndromes, Tumour Pathology and Molecular Testing. Histopathology 2023, 82, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Renault, A.L.; Mebirouk, N.; Fuhrmann, L.; Bataillon, G.; Cavaciuti, E.; Le Gal, D.; Girard, E.; Popova, T.; La Rosa, P.; Beauvallet, J.; et al. Morphology and Genomic Hallmarks of Breast Tumours Developed by ATM Deleterious Variant Carriers. Breast Cancer Res. 2018, 20, 28. [Google Scholar] [CrossRef] [Green Version]
- Mavaddat, N.; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Keeman, R.; Bolla, M.K.; Dennis, J.; Wang, Q.; Ahearn, T.U.; et al. Pathology of Tumors Associated with Pathogenic Germline Variants in 9 Breast Cancer Susceptibility Genes. JAMA Oncol. 2022, 8, e216744. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Fu, F.; Li, Z.; Feng, Q.; Wu, W.; Liu, Y.; Wang, C.; Chen, Y. Spectrum of PALB2 Germline Mutations and Characteristics of PALB2-Related Breast Cancer: Screening o;f 16,501 Unselected Patients with Breast Cancer and 5890 Controls by next-Generation Sequencing. Cancer 2020, 126, 3202–3208. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C DNA Change | Protein Change | Classification | rsID | |
|---|---|---|---|---|
| ATM (NM_000051.3) | c.6300C>G | p.(Tyr2100*) | PV | rs1591789955 |
| c.6300C>G | p.(Tyr2100*) | PV | rs1591789955 | |
| EXON 29 DELETION | LPV | Not applicable | ||
| c.2638+1G>A | LPV | Not referenced | ||
| CHEK2 (NM_007194.3) | c.1427C>T | p.(Thr476Met) | LPV | rs142763740 |
| c.793-1G>A | PV | rs730881687 | ||
| c.507del | p.(Phe169Leufs2*) | PV | rs587780183 | |
| c.470T>C | p.(Ile157Thr) | LPV | rs17879961 | |
| EXON 9 DELETION | LPV | Not applicable | ||
| PALB2 (NM_024675.3) | c.355C>T | p.(Gln119*) | PV | Not referenced |
| c.355C>T | p.(Gln119*) | PV | Not referenced | |
| RAD51D (NM_002878.3) | c.803G>A | p.(Trp268*) | PV | rs750219200 |
| c.803G>A | p.(Trp268*) | PV | rs750219200 | |
| c.757C>T | p.(Arg253*) | PV | rs137886232 | |
| RAD51C (NM_058216.2) | c.1026+5_1026+7delGTA | PV | rs587781410 |
| Characteristics | N° | % |
|---|---|---|
| Age at diagnosis | ||
| 21–40 | 4 | 26.7 |
| 41–60 | 9 | 60 |
| 61–80 | 2 | 13.3 |
| Histology | ||
| IDC-NST | 11 | 73.3 |
| INVASIVE MUCINOUS | 1 | 6.7 |
| INVASIVE LOBULAR | 2 | 13.3 |
| DUCTAL INTRAEPITHELIAL | 1 | 6.7 |
| HER2 profile | ||
| POSITIVE | 3 | 18.75 |
| NEGATIVE | 9 | 56.25 |
| UNKNOWN | 4 | 25 |
| ER profile | ||
| POSITIVE | 9 | 56.25 |
| NEGATIVE | 5 | 31.25 |
| UNKNOWN | 2 | 12.5 |
| PR profile | ||
| POSITIVE | 7 | 43.75 |
| NEGATIVE | 7 | 43.75 |
| UNKNOWN | 2 | 12.5 |
| Family history of BC | ||
| YES | 11 | 73.3 |
| NO | 4 | 26.7 |
| Family history of other cancer | ||
| YES | 8 | 53.3 |
| NO | 7 | 46.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turchiano, A.; Piglionica, M.; Martino, S.; Bagnulo, R.; Garganese, A.; De Luisi, A.; Chirulli, S.; Iacoviello, M.; Stasi, M.; Tabaku, O.; et al. Impact of High-to-Moderate Penetrance Genes on Genetic Testing: Looking over Breast Cancer. Genes 2023, 14, 1530. https://doi.org/10.3390/genes14081530
Turchiano A, Piglionica M, Martino S, Bagnulo R, Garganese A, De Luisi A, Chirulli S, Iacoviello M, Stasi M, Tabaku O, et al. Impact of High-to-Moderate Penetrance Genes on Genetic Testing: Looking over Breast Cancer. Genes. 2023; 14(8):1530. https://doi.org/10.3390/genes14081530
Chicago/Turabian StyleTurchiano, Antonella, Marilidia Piglionica, Stefania Martino, Rosanna Bagnulo, Antonella Garganese, Annunziata De Luisi, Stefania Chirulli, Matteo Iacoviello, Michele Stasi, Ornella Tabaku, and et al. 2023. "Impact of High-to-Moderate Penetrance Genes on Genetic Testing: Looking over Breast Cancer" Genes 14, no. 8: 1530. https://doi.org/10.3390/genes14081530
APA StyleTurchiano, A., Piglionica, M., Martino, S., Bagnulo, R., Garganese, A., De Luisi, A., Chirulli, S., Iacoviello, M., Stasi, M., Tabaku, O., Meneleo, E., Capurso, M., Crocetta, S., Lattarulo, S., Krylovska, Y., Lastella, P., Forleo, C., Stella, A., Bukvic, N., ... Resta, N. (2023). Impact of High-to-Moderate Penetrance Genes on Genetic Testing: Looking over Breast Cancer. Genes, 14(8), 1530. https://doi.org/10.3390/genes14081530