
Danon Disease: Entire LAMP2 Gene Deletion with Unusual Clinical Presentation—Case Report and Review of the Literature
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
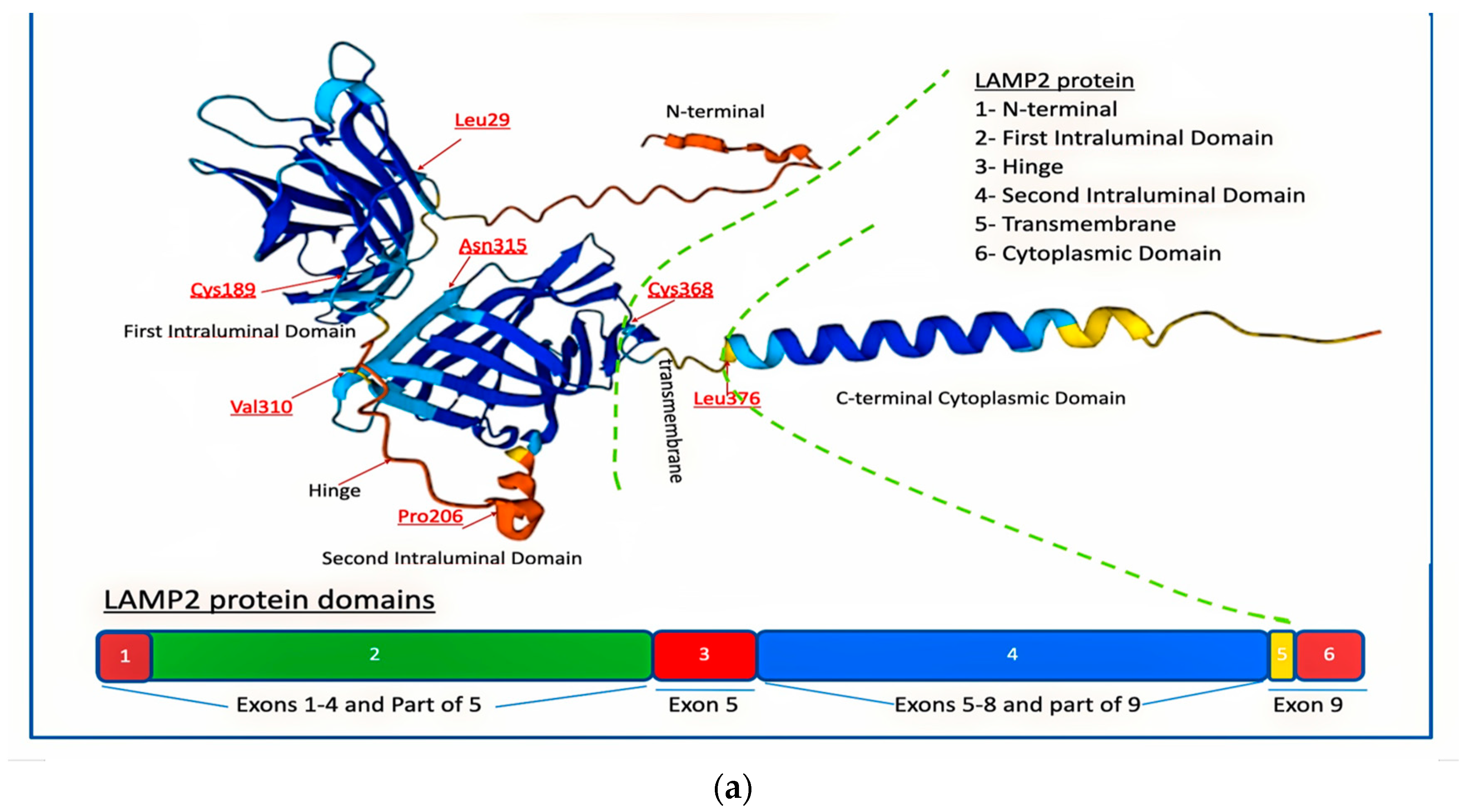
- The N-terminal cytoplasmic domain of LAMP2A is composed of approximately 110 amino acids and contains several protein–protein interaction motifs, such as a WW domain, a PDZ-binding motif, and a dileucine motif. These motifs enable LAMP2A to interact with other proteins and participate in various cellular signaling pathways, including autophagy.
- The transmembrane domain of LAMP2A consists of a single α-helix that is approximately 25 amino acids long. It spans the lysosomal membrane and anchors LAMP2A in place. The transmembrane domain is amphipathic, with hydrophobic residues on one side that interact with the lipid bilayer of the membrane [23].
- The luminal domain of LAMP2A that faces the inside of the lysosome is composed of approximately 150 amino acids and contains several glycosylation sites. Glycosylation is the process by which sugar molecules are attached to proteins, and it plays a crucial role in proper folding, stability, and function of LAMP2A. The luminal domain also contains several conserved cysteine residues that form disulfide bonds, which help stabilize the protein [23].
- The C-terminal cytoplasmic domain of LAMP2A faces the cytoplasm and is responsible for interactions with cytoskeletal proteins and lysosomal trafficking. It is composed of approximately 60 amino acids and contains a proline-rich motif and a dileucine motif. The proline-rich motif enables LAMP2A to interact with cytoskeletal proteins, such as actin, and participate in lysosomal trafficking. The dileucine motif is recognized by adaptor proteins that mediate the transport of LAMP2A from the trans-Golgi network to the lysosome [23].
2. Diagnostic Assessment—Methods and Results
Patient Evaluation
3. Whole-Exome Sequence
4. X Inactivation
5. Results
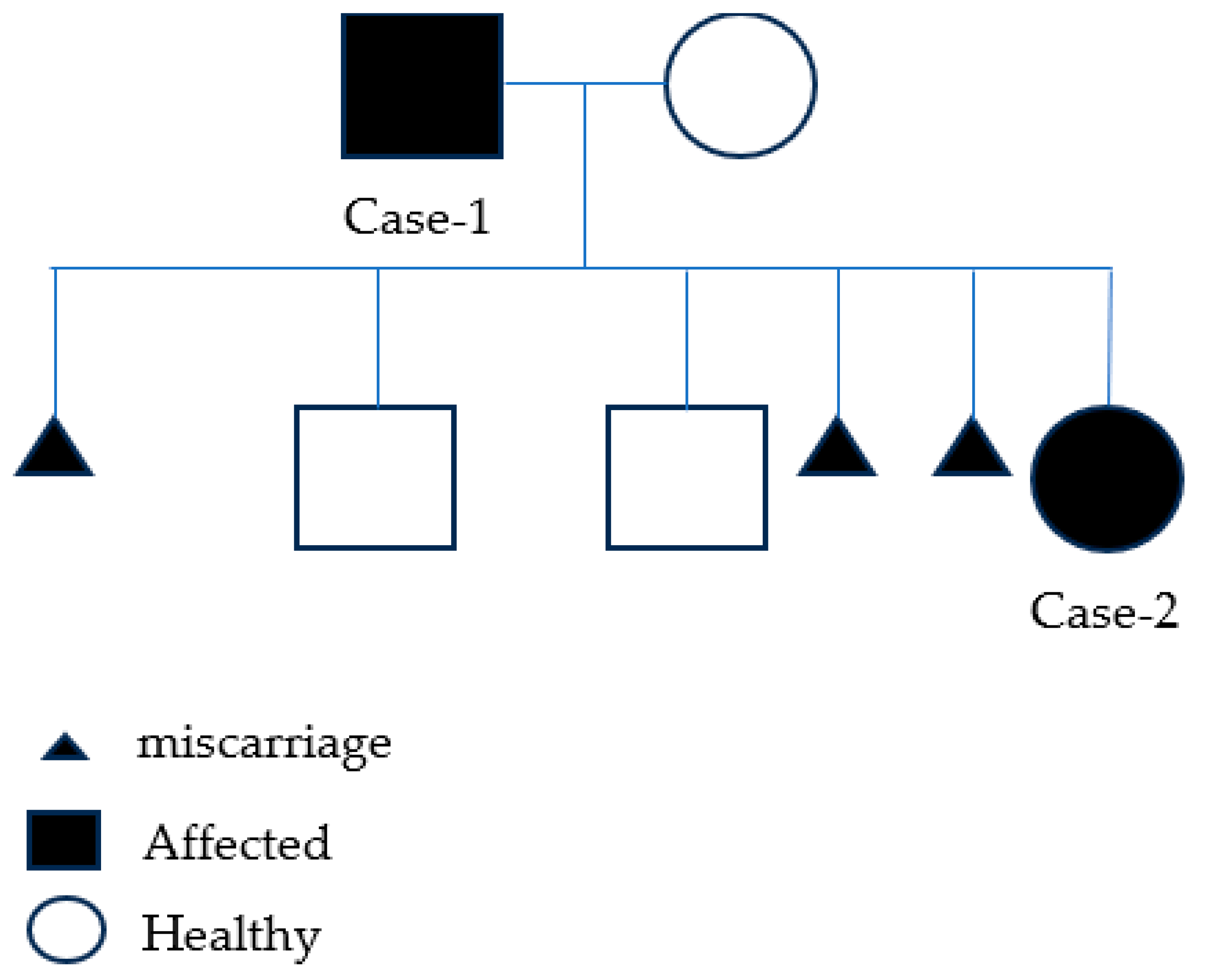
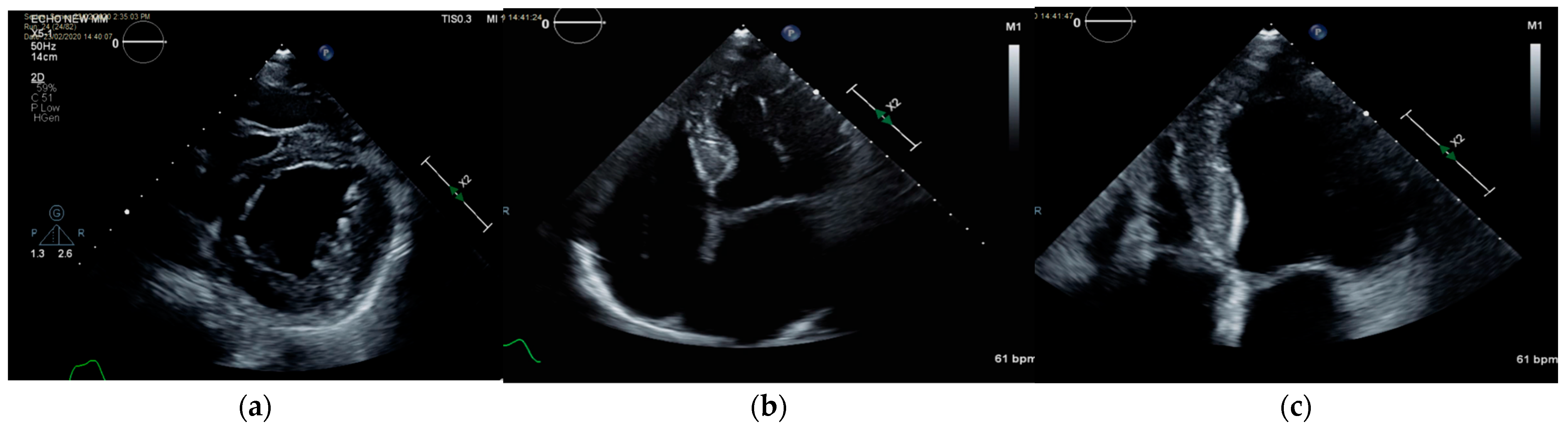
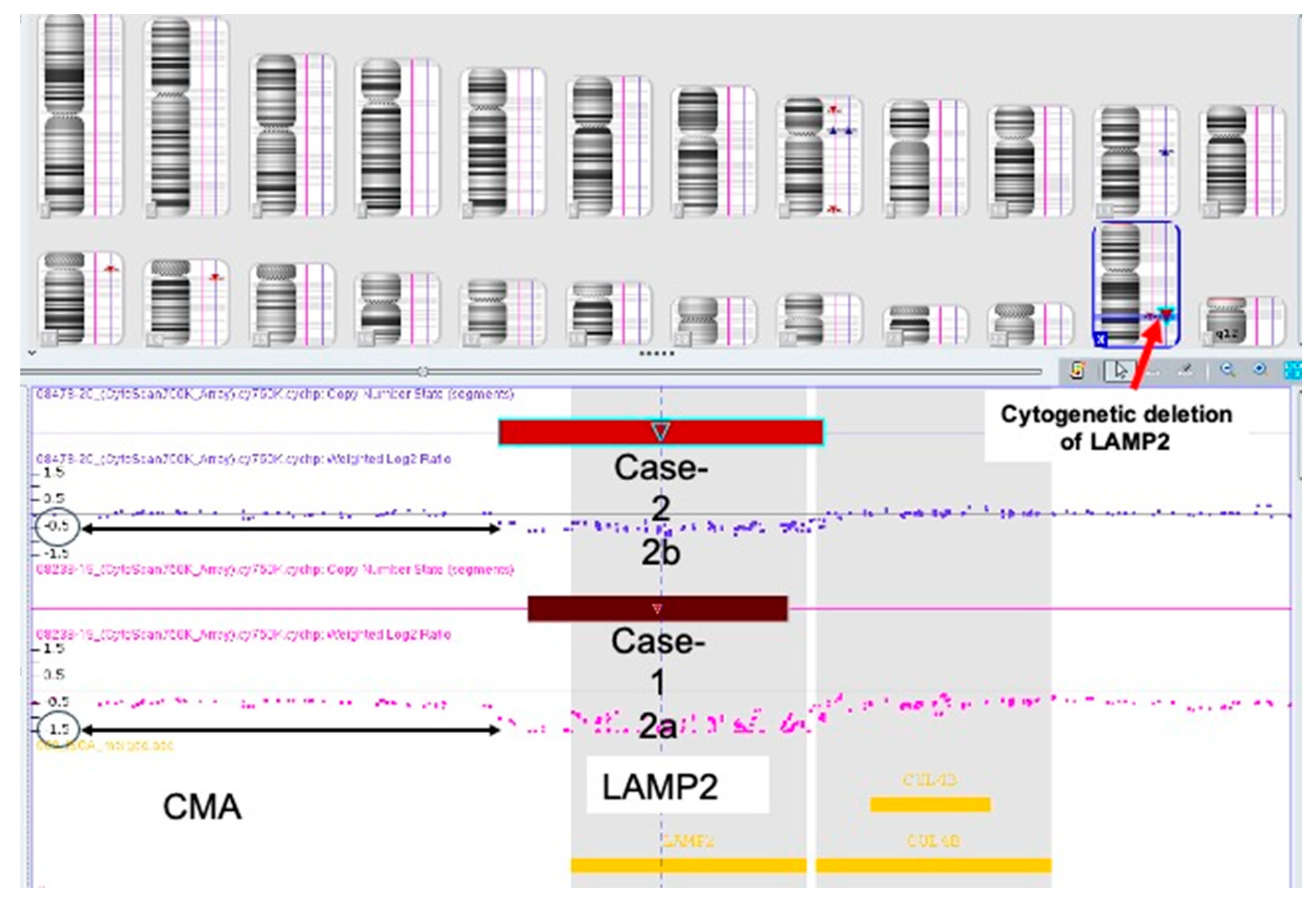
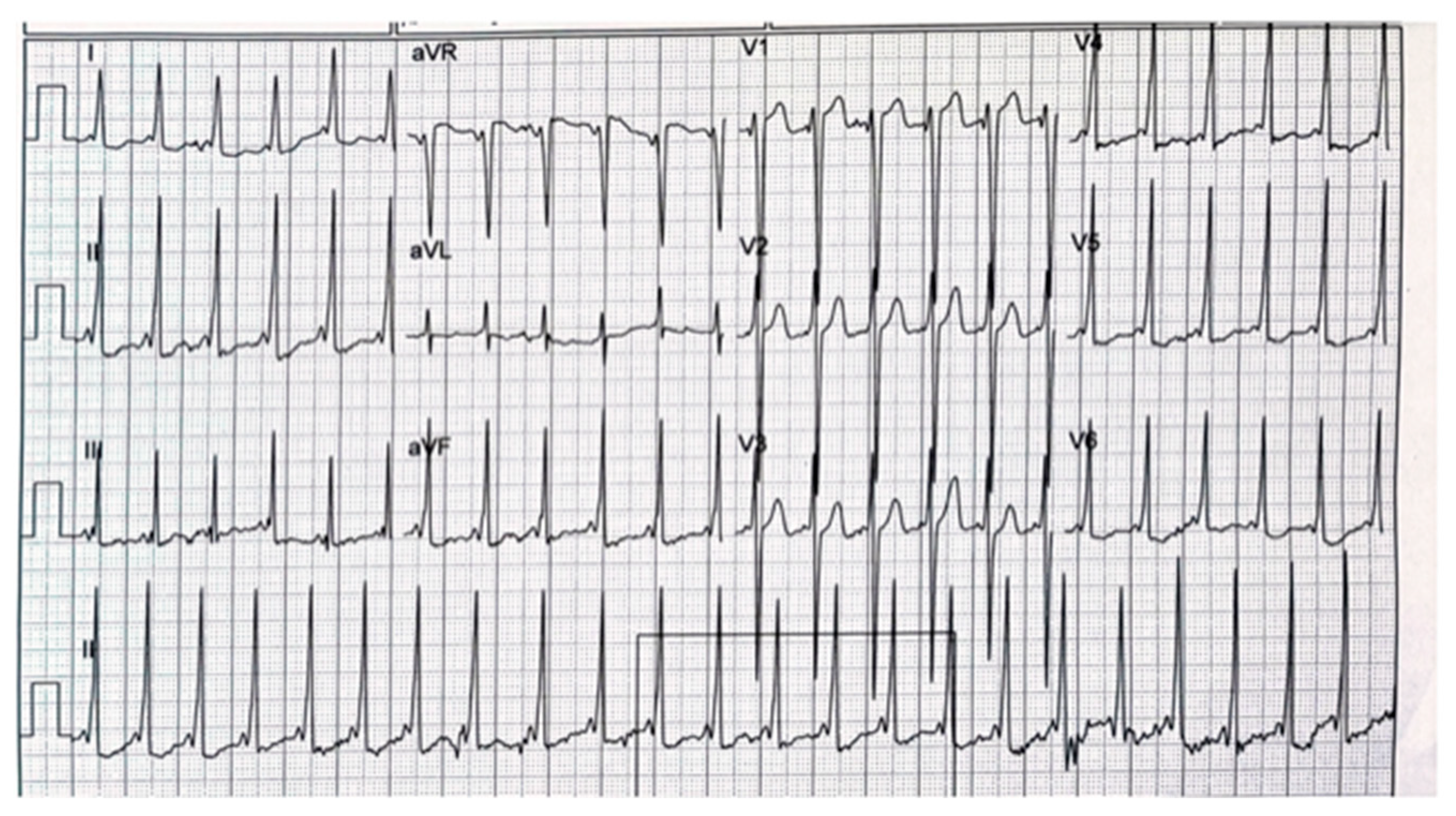
Case Description
6. X-Inactivation Study
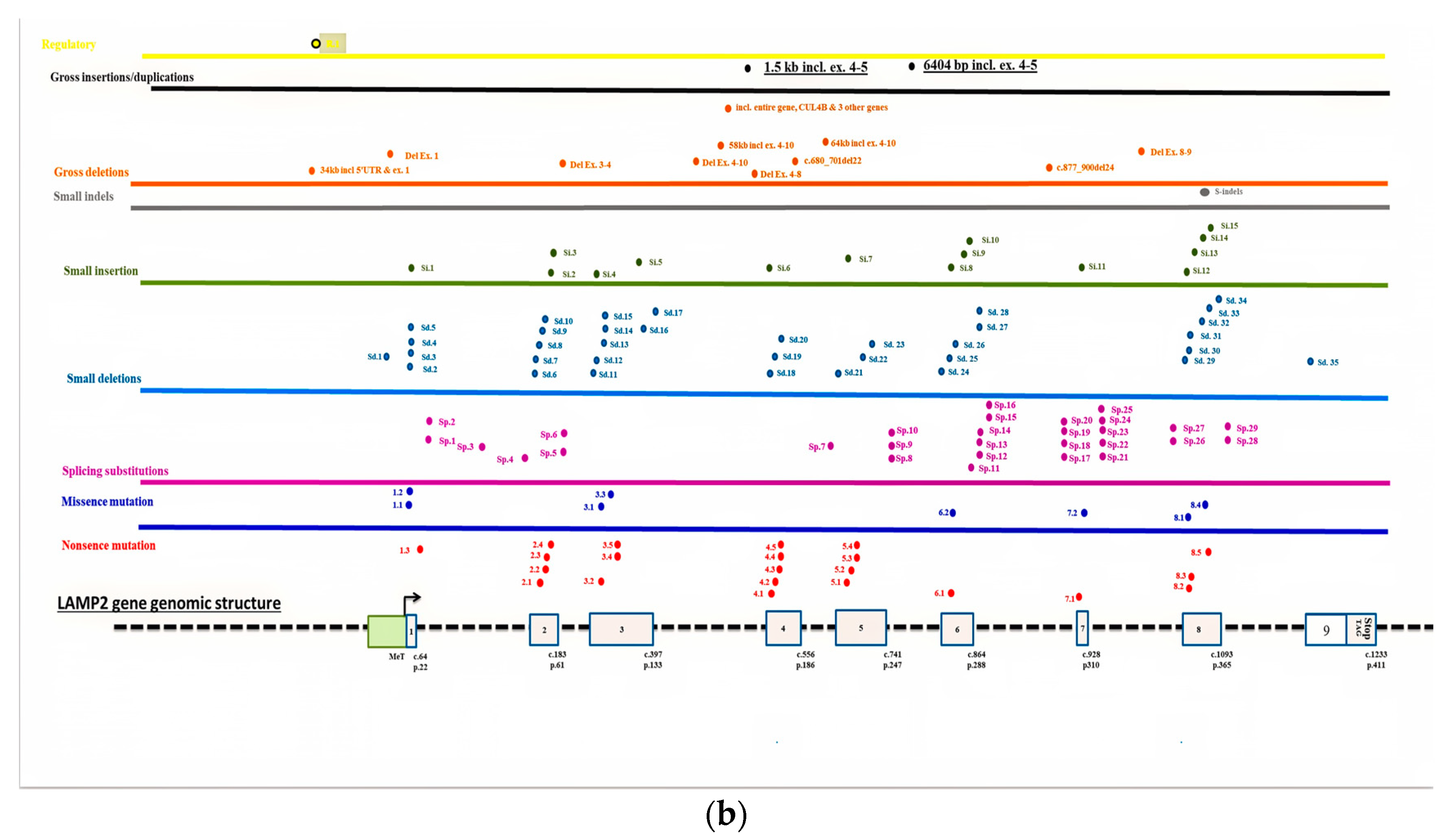
7. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cenacchi, G.; Papa, V.; Pegoraro, V.; Marozzo, R.; Fanin, M.; Angelini, C. Review: Danon Disease: Review of Natural History and Recent Advances. Neuropathol. Appl. Neurobiol. 2020, 46, 303–322. [Google Scholar] [CrossRef]
- Chi, C.; Leonard, A.; Knight, W.; Beussman, K.; Zhao, Y.; Cao, Y.; Londono, P.; Aune, E.; Trembley, M.; Smal, E.; et al. Lamp-2b Regulates Human Cardiomyocyte Function by Mediating Autophagosome-Lysosome Fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 Deficiency Causes X-Linked Vacuolar Cardiomyopathy and Myopathy (Danon Disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef]
- Ng, K.M.; Mok, P.Y.; Butler, A.W.; Ho, J.C.Y.; Choi, S.W.; Lee, Y.K.; Lai, W.H.; Au, K.W.; Lau, Y.M.; Wong, L.Y.; et al. Amelioration of X-Linked Related Autophagy Failure in Danon Disease with DNA Methylation Inhibitor. Circulation 2016, 134, 1373–1389. [Google Scholar] [CrossRef]
- Hashem, S.I.; Perry, C.N.; Bauer, M.; Han, S.; Clegg, S.D.; Ouyang, K.; Deacon, D.C.; Spinharney, M.; Panopoulos, A.D.; Izpisua Belmonte, J.C.; et al. Brief Report: Oxidative Stress Mediates Cardiomyocyte Apoptosis in a Human Model of Danon Disease and Heart Failure. Stem Cells 2015, 33, 2343–2350. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; McMahon, C.J.; Smith, L.R.; Bersola, J.; Adesina, A.M.; Breinholt, J.P.; Kearney, D.L.; Dreyer, W.J.; Denfield, S.W.; Price, J.F.; et al. Danon Disease as an Underrecognized Cause of Hypertrophic Cardiomyopathy in Children. Circulation 2005, 112, 1612–1617. [Google Scholar] [CrossRef] [Green Version]
- D’souza, R.S.; Levandowski, C.; Slavov, D.; Graw, S.L.; Allen, L.A.; Adler, E.; Mestroni, L.; Taylor, M.R.G. Danon Disease Clinical Features, Evaluation, and Management. Circ. Heart Fail. 2014, 7, 843–849. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, X.; Wang, F.; Liang, Y.; Deng, H.; Liao, H.; Zhang, Q.; Zhang, B.; Zhan, X.; Fang, X.; et al. Prevalence and Clinical Characteristics of Danon Disease among Patients with Left Ventricular Hypertrophy and Concomitant Electrocardiographic Preexcitation. Mol. Genet. Genom. Med. 2019, 7, e638. [Google Scholar] [CrossRef] [Green Version]
- Schorderet, D.F.; Cottet, S.; Lobrinus, J.A.; Borruat, F.X.; Balmer, A.; Munier, F.L. Retinopathy in Danon Disease. Arch. Ophthalmol. 2007, 125, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Prall, F.R.; Drack, A.; Taylor, M.; Ku, L.; Olson, J.L.; Gregory, D.; Mestroni, L.; Mandava, N. Ophthalmic Manifestations of Danon Disease. Ophthalmology 2006, 113, 1010–1013. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Liu, Y.; Zhang, X.; Niu, F.; Zheng, H.; Wang, L.; Kang, L.; Wang, K.; Xu, B. Danon Disease: A Case Report and Literature Review. Diagn. Pathol. 2021, 16, 39. [Google Scholar] [CrossRef]
- Brambatti, M.; Caspi, O.; Maolo, A.; Koshi, E.; Greenberg, B.; Taylor, M.R.G.; Adler, E.D. Danon Disease: Gender Differences in Presentation and Outcomes. Int. J. Cardiol. 2019, 286, 92–98. [Google Scholar] [CrossRef]
- Sugie, K.; Komaki, H.; Onoue, K.; Eura, N.; Shiota, T.; Tsukaguchi, H.; Namatame, S.; Koito, H.; Kiriyama, T.; Saito, Y.; et al. Clinicopathological Features and Management of Danon Disease in Japan: A Nationwide Survey. Neuromuscul. Disord. 2017, 27, S205. [Google Scholar] [CrossRef]
- Boucek, D.; Jirikowic, J.; Taylor, M. Natural History of Danon Disease. Genet. Med. 2011, 13, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Rowland, T.J.; Sweet, M.E.; Mestroni, L.; Taylor, M.R.G. Danon Disease—Dysregulation of Autophagy in a Multisystem Disorder with Cardiomyopathy. J. Cell Sci. 2016, 129, 2135–2143. [Google Scholar] [CrossRef] [Green Version]
- Sugie, K.; Nishino, I. Lysosomal Membrane Disorders: LAMP-2 Deficiency. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease: Fifth Edition; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Yardeni, M.; Weisman, O.; Mandel, H.; Weinberger, R.; Quarta, G.; Salazar-Mendiguchía, J.; Garcia-Pavia, P.; Lobato-Rodríguez, M.J.; Simon, L.F.; Dov, F.; et al. Psychiatric and Cognitive Characteristics of Individuals with Danon Disease (LAMP2 Gene Mutation). Am. J. Med. Genet. A 2017, 173, 2461–2466. [Google Scholar] [CrossRef]
- Tuneu, C.M.; Soria, V.; Gascón-Bayarri, J. Psychotic Symptoms in Danon Disease: A Clinical Case Report. Eur. Psychiatry 2021, 64, S736. [Google Scholar] [CrossRef]
- Miani, D.; Fresco, C.; Muser, D.; Driussi, M.; Nalli, C.; Tursi, V.; Livi, U.; Proclemer, A. Long Term Follow up after Heart Transplantation in Danon Disease. Eur. J. Heart Fail. 2015, 17, 244–245. [Google Scholar]
- Di Nora, C.; Miani, D.; D’Elia, A.V.; Poli, S.; Iascone, M.; Nucifora, G.; Finato, N.; Sponga, S.; Proclemer, A.; Livi, U. Heart Transplantation in Danon Disease: Long Term Single Centre Experience and Review of the Literature. Eur. J. Med. Genet. 2020, 63, 103645. [Google Scholar] [CrossRef]
- Salisbury, D.; Meredith, K. Neuropsychological Functioning Following Cardiac Transplant in Danon Disease. Dev. Neurorehabilit. 2019, 22, 67–70. [Google Scholar] [CrossRef]
- Taylor, M.R.G.; Adler, E.D. Danon Disease—GeneReviewsTM—NCBI Bookshelf. In GeneReviews®; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Ikami, Y.; Terasawa, K.; Watabe, T.; Yokoyama, S.; Hara-Yokoyama, M. The Two-Domain Architecture of LAMP2A within the Lysosomal Lumen Regulates Its Interaction with HSPA8/Hsc70. Autophagy Rep. 2022, 1, 205–209. [Google Scholar] [CrossRef]
- Endo, Y.; Furuta, A.; Nishino, I. Danon Disease: A Phenotypic Expression of LAMP-2 Deficiency. Acta Neuropathol. 2015, 129, 391–398. [Google Scholar] [CrossRef]
- Vatta, M.; Yang, Z.; Funke, B.H.; Cripe, L.H.; Vick, G.W.; Mancini-Dinardo, D.; Peña, L.S.; Kanter, R.J.; Wong, B.; Westerfield, B.H.; et al. LAMP2 Microdeletions in Patients With Danon Disease. Circ. Cardiovasc. Genet. 2010, 3, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI Sites near the Polymorphic CAG Repeat in the Human Androgen-Receptor Gene Correlates with X Chromosome Inactivation. Am. J. Hum. Genet. 1992, 51, 1229. [Google Scholar]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Corrigendum: Recommendations for Reporting of Secondary Findings in Clinical Exome and Genome Sequencing, 2016 Update (ACMG SF v2.0): A Policy Statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 484–485. [Google Scholar] [CrossRef] [Green Version]
- Majer, F.; Piherova, L.; Reboun, M.; Stara, V.; Pelak, O.; Norambuena, P.; Stranecky, V.; Krebsova, A.; Vlaskova, H.; Dvorakova, L.; et al. LAMP2 Exon-Copy Number Variations in Danon Disease Heterozygote Female Probands: Infrequent or Underdetected? Am. J. Med. Genet. A 2018, 176, 2430–2434. [Google Scholar] [CrossRef]
- Bottillo, I.; Giordano, C.; Cerbelli, B.; D’Angelantonio, D.; Lipari, M.; Polidori, T.; Majore, S.; Bertini, E.; D’Amico, A.; Giannarelli, D.; et al. A Novel LAMP2 Mutation Associated with Severe Cardiac Hypertrophy and Microvascular Remodeling in a Female with Danon Disease: A Case Report and Literature Review. Cardiovasc. Pathol. 2016, 25, 423–431. [Google Scholar] [CrossRef]
- Fu, L.; Luo, S.; Cai, S.; Hong, W.; Guo, Y.; Wu, J.; Liu, T.; Zhao, C.; Li, F.; Huang, H.; et al. Identification of LAMP2 Mutations in Early-Onset Danon Disease With Hypertrophic Cardiomyopathy by Targeted Next-Generation Sequencing. Am. J. Cardiol. 2016, 118, 888–894. [Google Scholar] [CrossRef]
- Dvornikov, A.V.; Wang, M.; Yang, J.; Zhu, P.; Le, T.; Lin, X.; Cao, H.; Xu, X. Phenotyping an Adult Zebrafish Lamp2 Cardiomyopathy Model Identifies MTOR Inhibition as a Candidate Therapy. J. Mol. Cell. Cardiol. 2019, 133, 199–208. [Google Scholar] [CrossRef]
- Manso, A.M.; Hashem, S.I.; Nelson, B.C.; Gault, E.; Soto-Hermida, A.; Villarruel, E.; Brambatti, M.; Bogomolovas, J.; Bushway, P.J.; Chen, C.; et al. Systemic AAV9.LAMP2B Injection Reverses Metabolic and Physiologic Multiorgan Dysfunction in a Murine Model of Danon Disease. Sci. Transl. Med. 2020, 12, eaax1744. [Google Scholar] [CrossRef]
- Řeboun, M.; Rybová, J.; Dobrovolný, R.; Včelák, J.; Veselková, T.; Štorkánová, G.; Mušálková, D.; Hřebíček, M.; Ledvinová, J.; Magner, M.; et al. X-Chromosome Inactivation Analysis in Different Cell Types and Induced Pluripotent Stem Cells Elucidates the Disease Mechanism in a Rare Case of Mucopolysaccharidosis Type Ll in a Female. Folia Biol. Czech Repub. 2016, 62, 82. [Google Scholar]
- Kingdom, R.; Wright, C.F. Incomplete Penetrance and Variable Expressivity: From Clinical Studies to Population Cohorts. Front. Genet. 2022, 13, 920390. [Google Scholar] [CrossRef]
- Majer, F.; Kousal, B.; Dusek, P.; Piherova, L.; Reboun, M.; Mihalova, R.; Gurka, J.; Krebsova, A.; Vlaskova, H.; Dvorakova, L.; et al. Alu-Mediated Xq24 Deletion Encompassing CUL4B, LAMP2, ATP1B4, TMEM255A, and ZBTB33 Genes Causes Danon Disease in a Female Patient. Am. J. Med. Genet. A 2020, 182, 219–223. [Google Scholar] [CrossRef]
- Zhang, J.; Chou, O.H.I.; Tse, Y.L.; Ng, K.M.; Tse, H.F. Application of Patient-Specific Ipscs for Modelling and Treatment of x-Linked Cardiomyopathies. Int. J. Mol. Sci. 2021, 22, 8132. [Google Scholar] [CrossRef]
- Chen, X.; Harting, J.; Farrow, E.; Thiffault, I.; Kasperaviciute, D.; Hoischen, A.; Gilissen, C.; Pastinen, T.; Eberle, M.A. Comprehensive SMN1 and SMN2 Profiling for Spinal Muscular Atrophy Analysis Using Long-Read PacBio HiFi Sequencing. Am. J. Hum. Genet. 2023, 110, 240–250. [Google Scholar] [CrossRef]
- Sanchez-Jimeno, C.; Blanco-Kelly, F.; López-Grondona, F.; Losada-Del Pozo, R.; Moreno, B.; Rodrigo-Moreno, M.; Martinez-Cayuelas, E.; Riveiro-Alvarez, R.; Fenollar-Cortés, M.; Ayuso, C.; et al. Attention Deficit Hyperactivity and Autism Spectrum Disorders as the Core Symptoms of AUTS2 Syndrome: Description of Five New Patients and Update of the Frequency of Manifestations and Genotype-Phenotype Correlation. Genes 2021, 12, 1360. [Google Scholar] [CrossRef]
- Kaygusuz, S.B.; Alavanda, C.; Kirkgoz, T.; Eltan, M.; Yavas Abali, Z.; Helvacioglu, D.; Guran, T.; Ata, P.; Bereket, A.; Turan, S. Does Genotype–Phenotype Correlation Exist in Vitamin D-Dependent Rickets Type IA: Report of 13 New Cases and Review of the Literature. Calcif. Tissue Int. 2021, 108, 576–586. [Google Scholar] [CrossRef]
- Pascual-Alonso, A.; Martínez-Monseny, A.F.; Xiol, C.; Armstrong, J. MECP2-Related Disorders in Males. Int. J. Mol. Sci. 2021, 22, 9610. [Google Scholar] [CrossRef]
- Gutiérrez-Cerrajero, C.; Sprecher, E.; Paller, A.S.; Akiyama, M.; Mazereeuw-Hautier, J.; Hernández-Martín, A.; González-Sarmiento, R. Ichthyosis. Nat. Rev. Dis. Primer 2023, 9, 2. [Google Scholar] [CrossRef]
- Fan, Y.; Xu, Y.; Shi, C. NOTCH2NLC-Related Disorders: The Widening Spectrum and Genotype–Phenotype Correlation. J. Med. Genet. 2022, 59, 1–9. [Google Scholar] [CrossRef]
- Mattei, M.G.; Matterson, J.; Chen, J.W.; Williams, M.A.; Fukuda, M. Two Human Lysosomal Membrane Glycoproteins, h-Lamp-1 and h-Lamp-2, Are Encoded by Genes Localized to Chromosome 13q34 and Chromosome Xq24-25, Respectively. J. Biol. Chem. 1990, 265, 7548–7551. [Google Scholar] [CrossRef]
- Sadikovic, B.; Aref-Eshghi, E.; Levy, M.A.; Rodenhiser, D. DNA Methylation Signatures in Mendelian Developmental Disorders as a Diagnostic Bridge between Genotype and Phenotype. Epigenomics 2019, 11, 563–575. [Google Scholar] [CrossRef]
- Lee, S.; Menzies, L.; Hay, E.; Ochoa, E.; Docquier, F.; Rodger, F.; Deshpande, C.; Foulds, N.C.; Jacquemont, S.; Jizi, K.; et al. Epigenotype-Genotype–Phenotype Correlations in SETD1A and SETD2 Chromatin Disorders. Hum. Mol. Genet. 2023, ddad079. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.-C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; Dubourg, C.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef]
- Dingemans, A.J.M.; Truijen, K.M.G.; van de Ven, S.; Bernier, R.; Bongers, E.M.H.F.; Bouman, A.; de Graaff—Herder, L.; Eichler, E.E.; Gerkes, E.H.; De Geus, C.M.; et al. The Phenotypic Spectrum and Genotype-Phenotype Correlations in 106 Patients with Variants in Major Autism Gene CHD8. Transl. Psychiatry 2022, 12, 421. [Google Scholar] [CrossRef]
- Foroutan, A.; Haghshenas, S.; Bhai, P.; Levy, M.A.; Kerkhof, J.; McConkey, H.; Niceta, M.; Ciolfi, A.; Pedace, L.; Miele, E.; et al. Clinical Utility of a Unique Genome-Wide DNA Methylation Signature for KMT2A-Related Syndrome. Int. J. Mol. Sci. 2022, 23, 1815. [Google Scholar] [CrossRef]
- van der Laan, L.; Rooney, K.; Trooster, T.M.; Mannens, M.M.; Sadikovic, B.; Henneman, P. DNA Methylation Episignatures: Insight into Copy Number Variation. Epigenomics 2022, 14, 1373–1388. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalata, A.; Bar-Shai, M.; Hadid, Y.; Mahroum, M.; Mintz, H.; Shalata, Z.E.; Radzishevsky, E.; Genizi, J.; Lorber, A.; Ben-Yosef, T.; et al. Danon Disease: Entire LAMP2 Gene Deletion with Unusual Clinical Presentation—Case Report and Review of the Literature. Genes 2023, 14, 1539. https://doi.org/10.3390/genes14081539
Shalata A, Bar-Shai M, Hadid Y, Mahroum M, Mintz H, Shalata ZE, Radzishevsky E, Genizi J, Lorber A, Ben-Yosef T, et al. Danon Disease: Entire LAMP2 Gene Deletion with Unusual Clinical Presentation—Case Report and Review of the Literature. Genes. 2023; 14(8):1539. https://doi.org/10.3390/genes14081539
Chicago/Turabian StyleShalata, Adel, Marina Bar-Shai, Yarin Hadid, Muhammad Mahroum, Hila Mintz, Zaher Eldin Shalata, Evgeny Radzishevsky, Jacob Genizi, Avraham Lorber, Tamar Ben-Yosef, and et al. 2023. "Danon Disease: Entire LAMP2 Gene Deletion with Unusual Clinical Presentation—Case Report and Review of the Literature" Genes 14, no. 8: 1539. https://doi.org/10.3390/genes14081539
APA StyleShalata, A., Bar-Shai, M., Hadid, Y., Mahroum, M., Mintz, H., Shalata, Z. E., Radzishevsky, E., Genizi, J., Lorber, A., Ben-Yosef, T., & Yaniv, L. (2023). Danon Disease: Entire LAMP2 Gene Deletion with Unusual Clinical Presentation—Case Report and Review of the Literature. Genes, 14(8), 1539. https://doi.org/10.3390/genes14081539