
First Record of Comparative Plastid Genome Analysis and Phylogenetic Relationships among Corylopsis Siebold & Zucc. (Hamamelidaceae)

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. Genome Assembly and Annotation
2.3. Comparative Plastid Genome Analysis of Corylopsis
2.4. Repeats and Nucleotide Diversity Analysis
2.5. Phylogenetic Analyses
3. Results
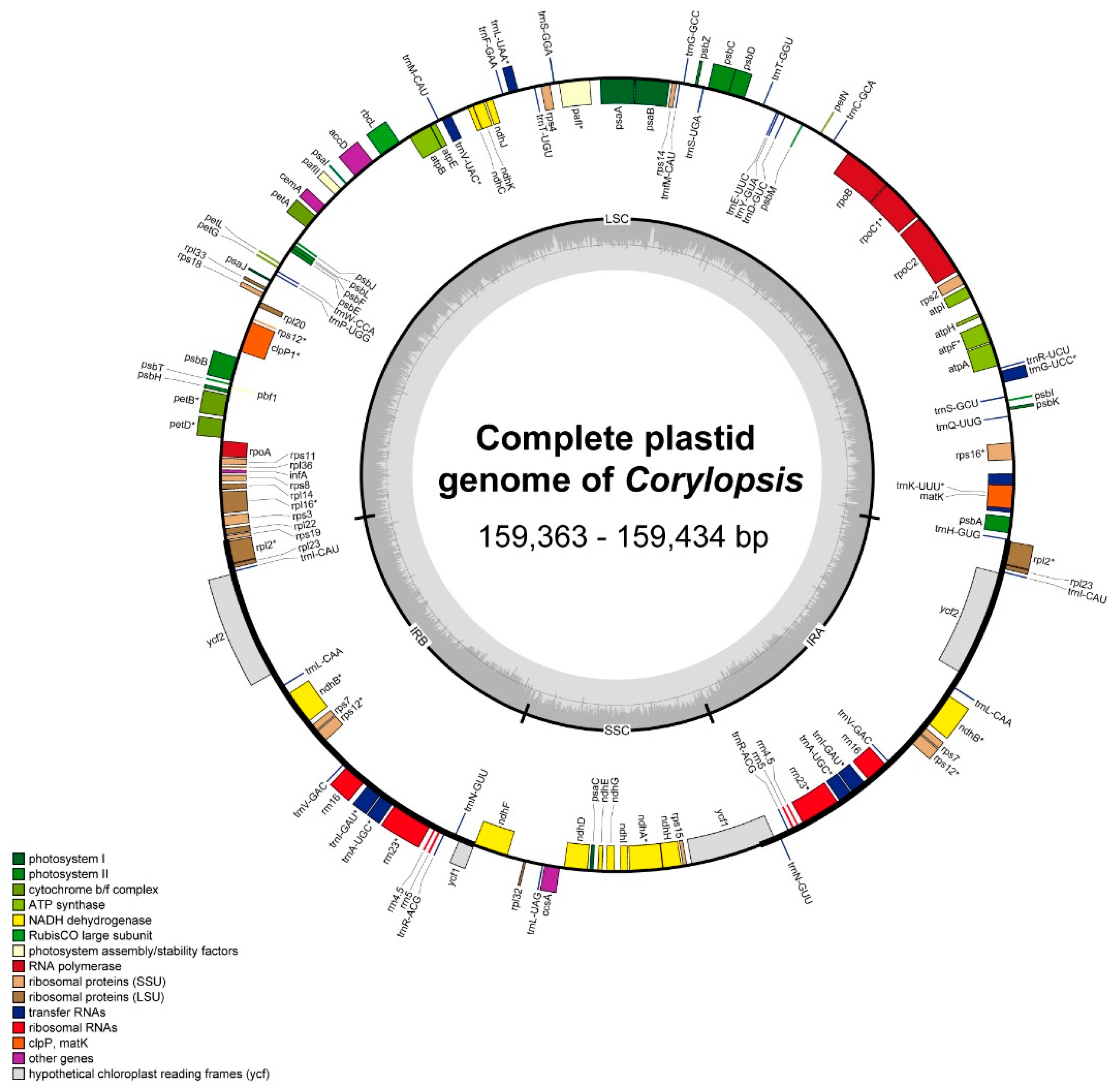
3.1. Plastid Genome Structure and Comparative Analysis of Corylopsis
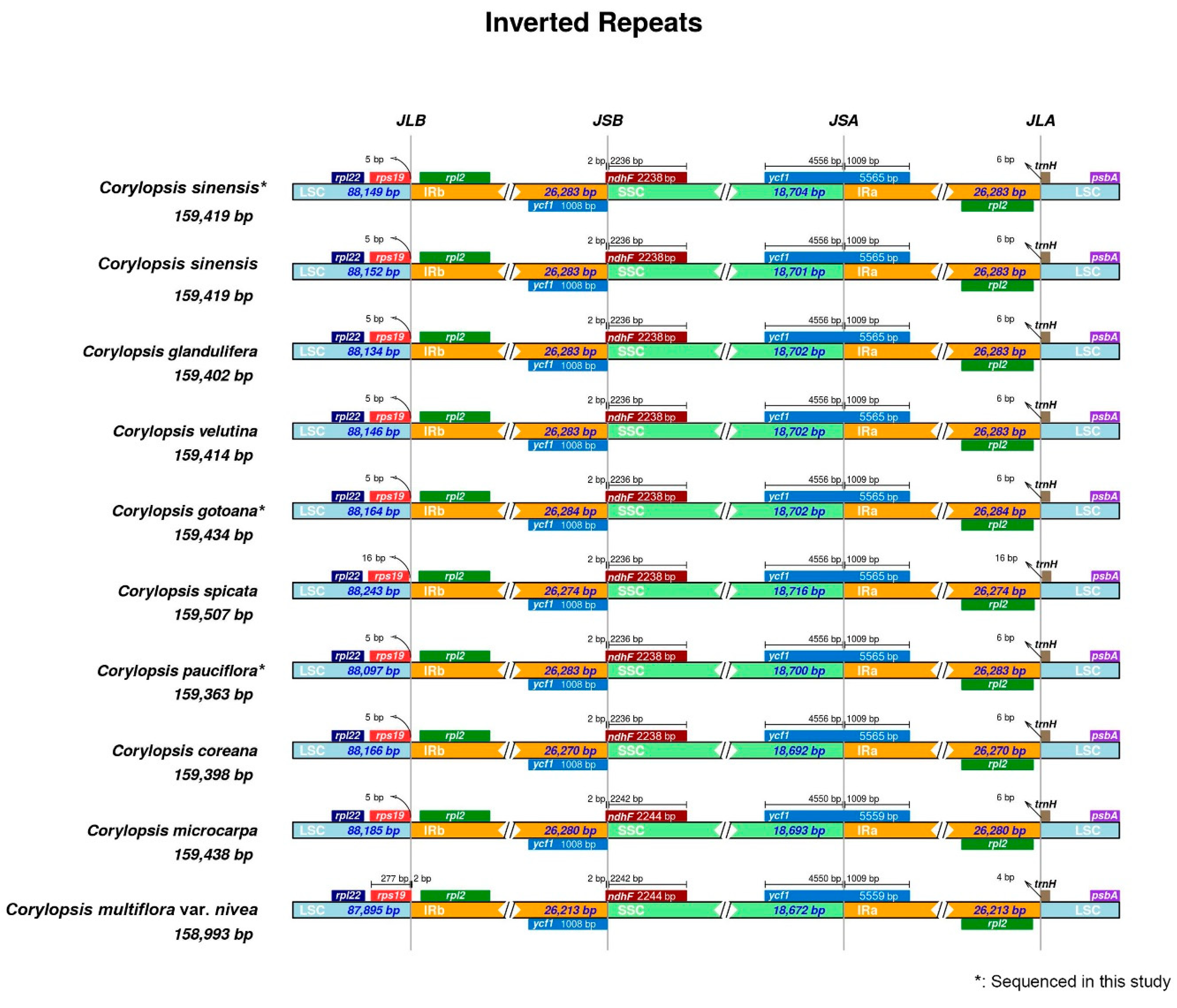
3.2. Repeat Analysis and Nucleotide Diversity Assessment
3.3. Phylogenetic Analyses of Corylopsis and Related Taxa
4. Discussion
4.1. Comparison of Plastid Genomes and Characteristics of Corylopsis
4.2. Divergence Hotspots of Corylopsis
4.3. Phylogenetic Relationships within Corylopsis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morley, B.; Chao, J.-M. A review of Corylopsis (Hamamelidaceae). J. Arnold Arbor. 1977, 58, 382–415. [Google Scholar] [CrossRef]
- Wu, Z.; Raven, P.H.; Hong, D. Pittosporaceae through Connaraceae. In Flora of China; Science Press: Beijing, China, 2003; Volume 9. [Google Scholar]
- Wang, N.; Chen, S.; Xie, L.; Wang, L.; Feng, Y.; Lv, T.; Fang, Y.; Ding, H. The complete chloroplast genomes of three Hamamelidaceae species: Comparative and phylogenetic analyses. Ecol. Evol. 2022, 12, e8637. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Ha, S.Y.; Oh, M.H.; Kim, H.H.; Kim, S.R.; Lee, M.W. Anti-oxidative and anti-proliferative activity on human prostate cancer cells lines of the phenolic compounds from Corylopsis coreana Uyeki. Molecules 2013, 18, 4876–4886. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Boo, H.O.; Ahn, T.; Bae, C.S. Protective effects of Erythronium japonicum and Corylopsis coreana Uyeki extracts against 1,3-dichloro-2-propanol-induced hepatotoxicity in rats. Appl. Microsc. 2020, 50, 29. [Google Scholar] [CrossRef]
- Li, C.; Chen, X.; Fang, D.; Li, G. A new bergenin derivative from Corylopsis willmottiae. Chem. Nat. Compd. 2011, 47, 194–196. [Google Scholar] [CrossRef]
- Yamanaka, M.; Kobayashi, S.; Setoguchi, H. Distinct geographical structure across species units evidenced by chloroplast DNA haplotypes and nuclear ribosomal ITS genotypes of Corylopsis (Hamamelidaceae) in the Japanese islands. Bot. J. Linn. Soc. 2008, 157, 501–518. [Google Scholar] [CrossRef]
- Barrett, R.D.H.; Hebert, P.D.N. Identifying spiders through DNA barcodes. Can. J. Zool. 2005, 83, 481–491. [Google Scholar] [CrossRef]
- Li, J.; Bogle, A.L.; Klein, A.S. Phylogenetic relationships in the Corylopsis complex (Hamamelidaceae): Evidence from sequences of the internal transcribed spacers of nuclear ribosomal DNA and morphology. Rhodora 1997, 99, 302–318. [Google Scholar]
- Li, J.; Bogle, A.L.; Klein, A.S. Phylogenetic relationships in the Hamamelidaceae: Evidence from the nucleotide sequences of the plastid genematK. Plant Syst. Evol. 1999, 218, 205–219. [Google Scholar] [CrossRef]
- Palmer, J.D.; Zamir, D. Chloroplast DNA evolution and phylogenetic relationships in Lycopersicon. Proc. Natl. Acad. Sci. USA 1982, 79, 5006–5010. [Google Scholar] [CrossRef]
- Pennington, R.T.; Lavin, M.; Ireland, H.; Klitgaard, B.; Preston, J.; Hu, J.M. Phylogenetic relationships of basal papilionoid legumes based upon sequences of the chloroplast trnL intron. Syst. Bot. 2001, 26, 537–556. [Google Scholar]
- Dobrogojski, J.; Adamiec, M.; Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant 2020, 42, 98. [Google Scholar] [CrossRef]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M. The chloroplast genome. In 10 Years Plant Molecular Biology; Springer: Dordrecht, The Netherlands, 1992; pp. 149–168. [Google Scholar]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef]
- Jones, S.S.; Burke, S.V.; Duvall, M.R. Phylogenomics, molecular evolution, and estimated ages of lineages from the deep phylogeny of Poaceae. Plant Syst. Evol. 2014, 300, 1421–1436. [Google Scholar] [CrossRef]
- Kim, T.H.; Kim, J.H. Molecular phylogeny and historical biogeography of Goodyera R. Br. (Orchidaceae): A case of the vicariance between East Asia and North America. Front. Plant Sci. 2022, 13, 850170. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.S.; Ha, Y.-H.; Jeong, K.S.; Joo, M.; Chang, K.S.; Choi, K. The complete chloroplast genome of Corylopsis coreana (Hamamelidaceae). Conserv. Genet. Resour. 2019, 11, 291–293. [Google Scholar] [CrossRef]
- Kim, C.; Kim, S.C.; Kim, J.H. Historical biogeography of Melanthiaceae: A case of out-of-North America through the Bering land bridge. Front. Plant Sci. 2019, 10, 396. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Shin, S.; Ahn, J.Y.; Lee, J.W. Complete chloroplast genome of Corylopsis spicata and phylogenetic analysis. Mitochondrial DNA B Resour. 2019, 4, 2700–2701. [Google Scholar] [CrossRef] [PubMed]
- Lv, T.; Chen, S.; Zhao, R.; Wang, N.; Fang, Y. The complete chloroplast genome sequence of Corylopsis multiflora Hance var. nivea Chang. Mitochondrial DNA B Resour. 2021, 6, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gu, J.; Chang, H. The complete chloroplast genome sequence of Corylopsis sinensis (Hamamelidaceae). Mitochondrial DNA B Resour. 2022, 7, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; NISC Comparative Sequencing Program; Green, E.D.; Sidow, A.; Batzoglou, S. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. S2), W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics, and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP: Phylogenetic Analysis Using Parsimony, Mac Version 3.1.1; Illinois Natural History Survey: Champaign, IL, USA, 1993. [Google Scholar]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Guo, X.; Liu, Q.; Wang, K. Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PLoS ONE 2019, 14, e0216645. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2016, 7, 2054. [Google Scholar] [CrossRef]
- Menezes, A.P.A.; Resende-Moreira, L.C.; Buzatti, R.S.O.; Nazareno, A.G.; Carlsen, M.; Lobo, F.P.; Kalapothakis, E.; Lovato, M.B. Chloroplast genomes of Byrsonima species (Malpighiaceae): Comparative analysis and screening of high divergence sequences. Sci. Rep. 2018, 8, 2210. [Google Scholar] [CrossRef]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef]
- Palmer, J.D.; Thompson, W.F. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 1982, 29, 537–550. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Guda, C.; Lee, S.B.; Daniell, H. Stable expression of a biodegradable protein-based polymer in tobacco chloroplasts. Plant Cell Rep. 2000, 19, 257–262. [Google Scholar] [CrossRef]
- Wu, L.; Nie, L.; Wang, Q.; Xu, Z.; Wang, Y.; He, C.; Song, J.; Yao, H. Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae. Sci. Rep. 2021, 11, 14643. [Google Scholar] [CrossRef]
- Lee, H.R.; Kim, K.A.; Kim, B.Y.; Park, Y.J.; Lee, Y.B.; Cheon, K.S. The complete chloroplast genome sequences of eight Orostachys species: Comparative analysis and assessment of phylogenetic relationships. PLoS ONE 2022, 17, e0277486. [Google Scholar] [CrossRef]
- Li, L.; Hu, Y.; He, M.; Zhang, B.; Wu, W.; Cai, P.; Huo, D.; Hong, Y. Comparative chloroplast genomes: Insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genom. 2021, 22, 138. [Google Scholar] [CrossRef]
- Plunkett, G.M.; Downie, S.R. Expansion and contraction of the chloroplast inverted repeat in Apiaceae subfamily Apioideae. Syst. Bot. 2000, 25, 648–667. [Google Scholar] [CrossRef]
- Peng, J.-Y.; Zhang, X.-S.; Zhang, D.-G.; Wang, Y.; Deng, T.; Huang, X.-H.; Kuang, T.-H.; Zhou, Q. Newly reported chloroplast genome of Sinosenecio albonervius Y. Liu & QE Yang and comparative analyses with other Sinosenecio species. BMC Genom. 2022, 23, 639. [Google Scholar] [CrossRef]
- Sharp, P.M.; Matassi, G. Codon usage and genome evolution. Curr. Opin. Genet. Dev. 1994, 4, 851–860. [Google Scholar] [CrossRef]
- Carlini, D.B.; Stephan, W. In vivo introduction of unpreferred synonymous codons into the Drosophila Adh gene results in reduced levels of ADH protein. Genetics 2003, 163, 239–243. [Google Scholar] [CrossRef]
- Hiraoka, Y.; Kawamata, K.; Haraguchi, T.; Chikashige, Y. Codon usage bias is correlated with gene expression levels in the fission yeast Schizosaccharomyces pombe. Genes. Cells 2009, 14, 499–509. [Google Scholar] [CrossRef]
- Romero, H.; Zavala, A.; Musto, H. Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 2000, 28, 2084–2090. [Google Scholar] [CrossRef]
- Romero, H.; Zavala, A.; Musto, H.; Bernardi, G. The influence of translational selection on codon usage in fishes from the family Cyprinidae. Gene 2003, 317, 141–147. [Google Scholar] [CrossRef]
- Gao, B.; Yuan, L.; Tang, T.; Hou, J.; Pan, K.; Wei, N. The complete chloroplast genome sequence of Alpinia oxyphylla Miq. and comparison analysis within the Zingiberaceae family. PLoS ONE 2019, 14, e0218817. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, Y.; Tng, D.Y.; Zhou, J.; Zhang, Y.; Wang, Z.; Li, P.; Wang, Z. Comparative chloroplast genomics of Litsea Lam. (Lauraceae) and its phylogenetic implications. Forests 2021, 12, 744. [Google Scholar] [CrossRef]
- Xue, J.; Wang, S.; Zhou, S.L. Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am. J. Bot. 2012, 99, e240–e244. [Google Scholar] [CrossRef]
- Yang, A.H.; Zhang, J.J.; Yao, X.H.; Huang, H.W. Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 2011, 98, e123–e126. [Google Scholar] [CrossRef]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef]
- Song, Y.; Chen, Y.; Lv, J.; Xu, J.; Zhu, S.; Li, M. Comparative chloroplast genomes of Sorghum species: Sequence divergence and phylogenetic relationships. BioMed Res. Int. 2019, 2019, 5046958. [Google Scholar] [CrossRef]
- Wang, L.; Wuyun, T.-N.; Du, H.; Wang, D.; Cao, D. Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genet. Genomes 2016, 12, 12. [Google Scholar] [CrossRef]
- Park, I.; Yang, S.; Choi, G.; Kim, W.J.; Moon, B.C. The complete chloroplast genome sequences of Aconitum pseudolaeve and Aconitum longecassidatum, and development of molecular markers for distinguishing species in the Aconitum subgenus Lycoctonum. Molecules 2017, 22, 2012. [Google Scholar] [CrossRef]
- Jung, J.; Kim, C.; Kim, J.H. Insights into phylogenetic relationships and genome evolution of subfamily Commelinoideae (Commelinaceae Mirb.) inferred from complete chloroplast genomes. BMC Genom. 2021, 22, 231. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, Y.; Liu, Y.M.; Xie, D.F.; He, X.J.; Zhou, S.D. Comparative chloroplast genomics of Fritillaria (Liliaceae), inferences for phylogenetic relationships between Fritillaria and Lilium and plastome evolution. Plants 2020, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Terakami, S.; Matsumura, Y.; Kurita, K.; Kanamori, H.; Katayose, Y.; Yamamoto, T.; Katayama, H. Complete sequence of the chloroplast genome from pear (Pyrus pyrifolia): Genome structure and comparative analysis. Tree Genet. Genomes 2012, 8, 841–854. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef]
- Shi, S.; Chang, H.T.; Chen, Y.; Qu, L.; Wen, J. Phylogeny of the Hamamelidaceae based on the ITS sequences of nuclear ribosomal DNA. Biochem. Syst. Ecol. 1998, 26, 55–69. [Google Scholar] [CrossRef]
- Bobrov, A.V.; Roslov, M.S.; Romanov, M.S. Phylogenetic biogeography of Hamamelidaceae s. l. based on molecular data. Vestn. St. Petersbg Univ. Earth Sci. 2020, 65, 224–244. [Google Scholar] [CrossRef]
- Xiao, M.; Li, Y.; Xiong, X.; Shen, S.; Song, J.; Liu, Q.; Jiang, T.; Luo, X.; Zhang, L. Complete chloroplast genome of Loropetalum chinense var. rubrum and phylogenetic analysis from Baoshan, China. Mitochondrial DNA Part B 2021, 6, 3336–3337. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Xiang, K.; Ortiz, R.D.C.; Jabbour, F.; Wang, W. Integrating palaeontological and molecular data uncovers multiple ancient and recent dispersals in the pantropical Hamamelidaceae. J. Biogeogr. 2019, 46, 2622–2631. [Google Scholar] [CrossRef]
- Chang, C.; Chang, K. Typification of Corylopsis coreana (Hamamelidaceae) and Carpinus laxiflora var. longispica (Betulaceae). J. Jpn. Bot. 2010, 85, 270–276. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Length (bp) and GC Content (%) | GenBank Accession Number | |||
|---|---|---|---|---|---|
| LSC | SSC | IR | Total | ||
| Corylopsis sinensis * | 88,149 (36.1%) | 18,704 (32.7%) | 26,283 (43.1%) | 159,419 (38.0%) | PP273282 |
| Corylopsis sinensis | 88,152 (36.1%) | 18,701 (32.7%) | 26,283 (43.1%) | 159,419 (38.0%) | MZ590567 |
| Corylopsis glandulifera | 88,134 (36.1%) | 18,702 (32.6%) | 26,283 (43.1%) | 159,402 (38.0%) | MZ642354 |
| Corylopsis velutina | 88,146 (36.1%) | 18,702 (32.7%) | 26,283 (43.1%) | 159,414 (38.0%) | MZ823391 |
| Corylopsis gotoana * | 88,164 (36.1%) | 18,702 (32.7%) | 26,284 (43.1%) | 159,434 (38.0%) | PP273280 |
| Corylopsis spicata | 88,243 (36.1%) | 18,716 (32.7%) | 26,274 (43.1%) | 159,507 (38.0%) | MK942341 |
| Corylopsis pauciflora * | 88,097 (36.1%) | 18,700 (32.7%) | 26,283 (43.1%) | 159,363 (38.0%) | PP273281 |
| Corylopsis coreana | 88,166 (36.1%) | 18,692 (32.7%) | 26,270 (43.1%) | 159,398 (38.0%) | MG835449 |
| Corylopsis microcarpa | 88,185 (36.1%) | 18,693 (32.6%) | 26,280 (43.1%) | 159,438 (38.0%) | MZ642356 |
| Corylopsis multiflora var. nivea | 87,895 (36.1%) | 18,672 (32.6%) | 26,213 (43.1%) | 158,993 (38.0%) | MW043717 |
| Loropetalum chinense | 88,160 (36.1%) | 18,770 (32.7%) | 26,257 (43.1%) | 159,444 (38.0%) | NC_060831 |
| Loropetalum subcordatum | 88,216 (36.1%) | 18,494 (32.7%) | 25,998 (43.1%) | 158,706 (38.0%) | NC_037694 |
| Distylium myricoides | 87,847 (36.2%) | 18,780 (32.5%) | 26,233 (43.1%) | 159,093 (38.0%) | NC_059883 |
| Distylium racemosum | 87,863 (36.2%) | 18,782 (32.5%) | 26,231 (43.1%) | 159,107 (38.0%) | NC_059886 |
| Hamamelis mollis | 88,301 (36.1%) | 18,762 (32.5%) | 26,334 (43.1%) | 159,731 (38.0%) | NC_037881 |
| Sinowilsonia henryi | 87,507 (36.4%) | 18,768 (32.8%) | 26,233 (43.1%) | 158,741 (38.2%) | NC_036069 |
| Mytilaria laosensis | 89,016 (35.9%) | 18,127 (32.8%) | 26,399 (43.1%) | 159,941 (37.9%) | NC_048997 |
| Rhodoleia championii | 88,144 (35.8%) | 18,131 (32.3%) | 26,420 (42.9%) | 159,115 (37.7%) | NC_045276 |
| Liquidambar styraciflua | 88,891 (36.1%) | 18,977 (32.4%) | 26,441 (43.0%) | 160,750 (37.9%) | NC_046938 |
| Liquidambar orientalis | 88,882 (36.1%) | 18,947 (32.4%) | 26,471 (43.1%) | 160,771 (37.9%) | NC_046937 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, T.-H.; Ha, Y.-H.; Setoguchi, H.; Choi, K.; Kim, S.-C.; Kim, H.-J. First Record of Comparative Plastid Genome Analysis and Phylogenetic Relationships among Corylopsis Siebold & Zucc. (Hamamelidaceae). Genes 2024, 15, 380. https://doi.org/10.3390/genes15030380
Kim T-H, Ha Y-H, Setoguchi H, Choi K, Kim S-C, Kim H-J. First Record of Comparative Plastid Genome Analysis and Phylogenetic Relationships among Corylopsis Siebold & Zucc. (Hamamelidaceae). Genes. 2024; 15(3):380. https://doi.org/10.3390/genes15030380
Chicago/Turabian StyleKim, Tae-Hee, Young-Ho Ha, Hiroaki Setoguchi, Kyung Choi, Sang-Chul Kim, and Hyuk-Jin Kim. 2024. "First Record of Comparative Plastid Genome Analysis and Phylogenetic Relationships among Corylopsis Siebold & Zucc. (Hamamelidaceae)" Genes 15, no. 3: 380. https://doi.org/10.3390/genes15030380
APA StyleKim, T. -H., Ha, Y. -H., Setoguchi, H., Choi, K., Kim, S. -C., & Kim, H. -J. (2024). First Record of Comparative Plastid Genome Analysis and Phylogenetic Relationships among Corylopsis Siebold & Zucc. (Hamamelidaceae). Genes, 15(3), 380. https://doi.org/10.3390/genes15030380