
Principles in the Management of Glioblastoma

 ,
,
Abstract
:1. Introduction
2. Epidemiology
3. Classification of Glioblastoma
- −
- Glioblastoma, isocitrate dehydrogenase (IDH) wildtype (90% of cases), developing de novo at about 60 years of age;
- −
- Glioblastoma, IDH-mutant (10% of cases), secondary glioblastoma that usually develops in younger patients with gliomas of higher differentiation (WHO grades I–III); it carries a significantly better prognosis than wildtype IDH [17];
- −
- Glioblastoma not otherwise specified (NOS), the IDH mutation status could not be determined due to a lack of histological or molecular material for testing;
- −
- Not-elsewhere-classified (NEC) glioblastoma, fourth category distinguished in recent years.
- adult patient
- diffuse astrocytic tumour
- IDH-wildtype
- and at least one of the following:
- ○
- necrosis
- ○
- microvascular proliferation
- ○
- TERT promoter mutation
- ○
- EGFR gene amplification
- ○
- combined gain of whole chromosome 7 and loss of chromosome 10 [+7/−10].
4. Molecular Pathogenesis of Glioblastoma
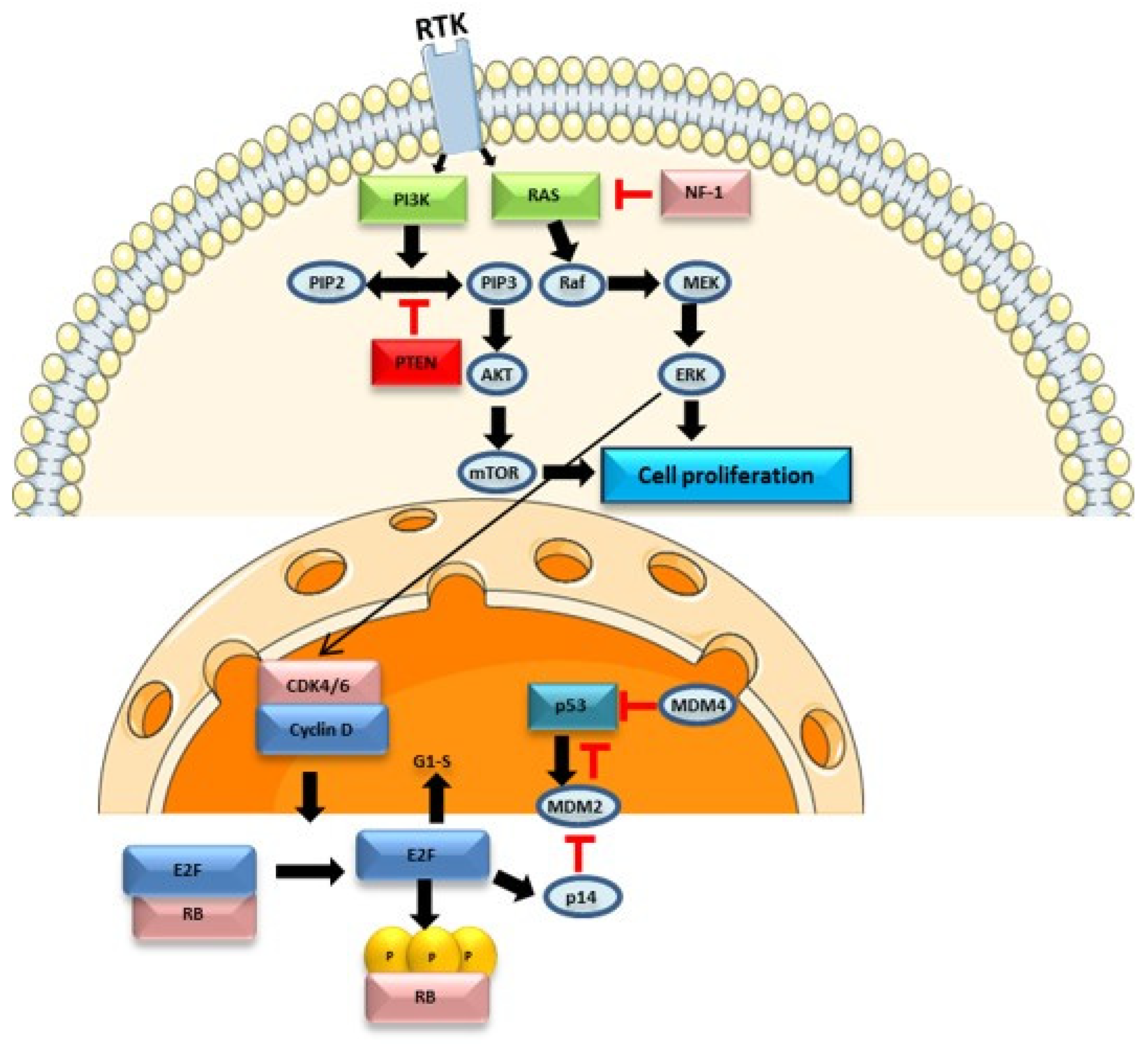
4.1. Cell Signalling Pathways in Glioblastoma
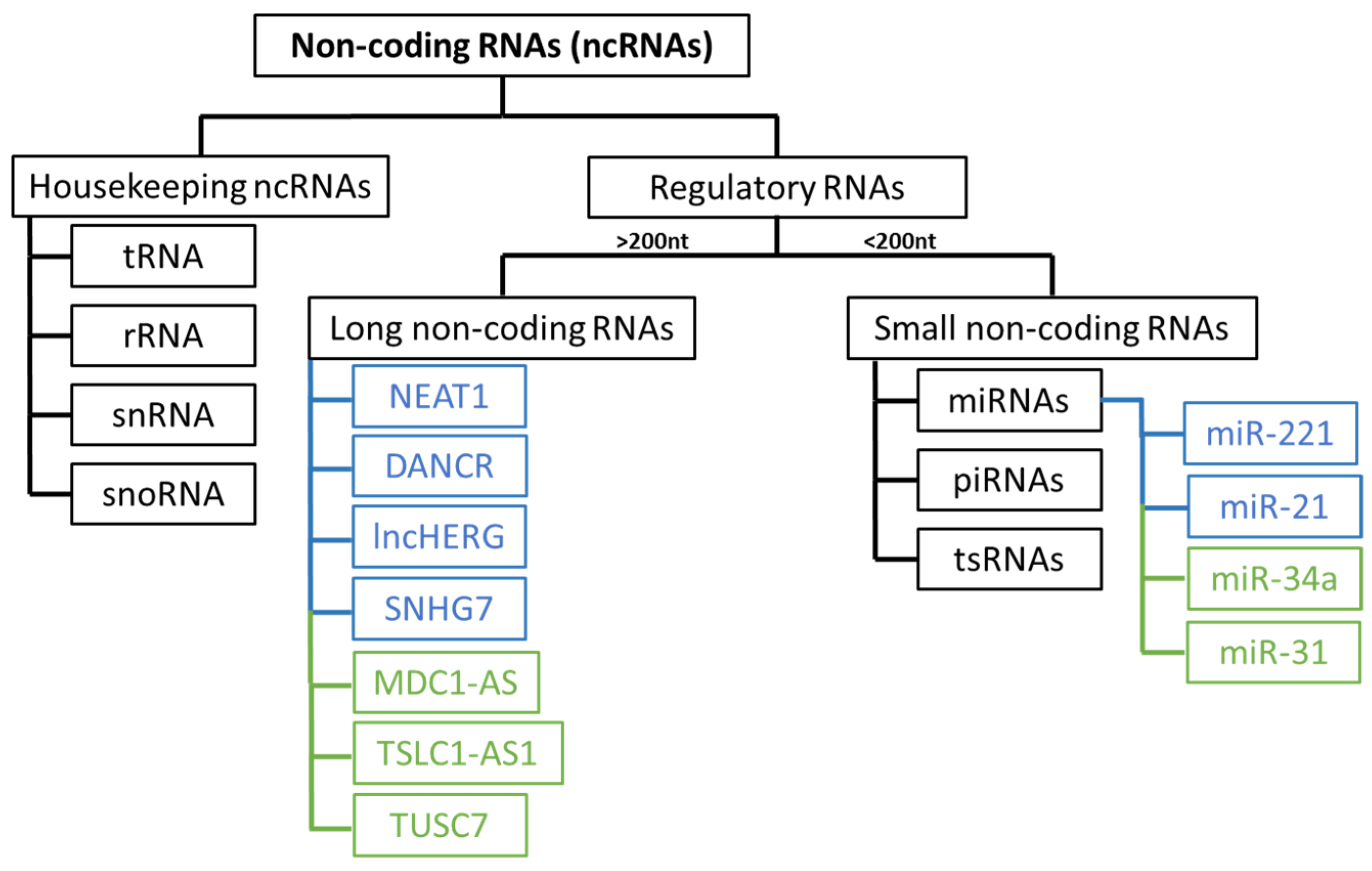
4.2. Epigenetic Mechanisms
4.3. Multi-Omics Approach
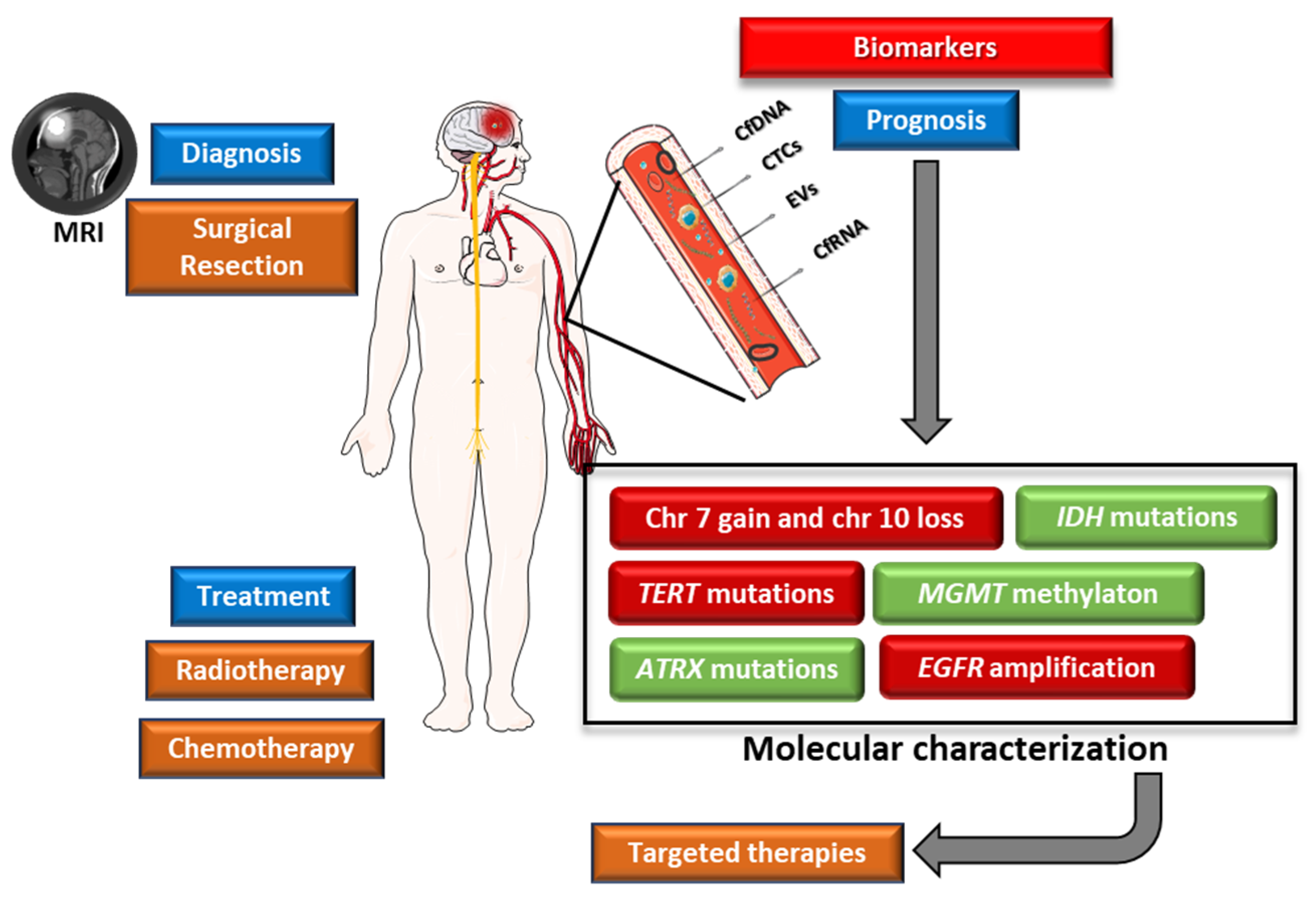
5. Prognostic Biomarkers
5.1. IDH1 and IDH2 Mutational Status
5.2. Alterations in ATRX (ATRX Chromatin Remodeler)
5.3. Alterations in TERT (Telomerase Reverse Transcriptase)
5.4. Alterations in CDKN2A (Cyclin Dependent Kinase Inhibitor 2A)
5.5. 1p/19q Codeletion
5.6. Chromosome 7 Gain and Chromosome 10 Loss
5.7. EGFR Mutations
5.8. MGMT Promoter Methylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Classification | Effect | References |
|---|---|---|---|
| IDH1 and IDH2 | Prognostic | Mutations in IDH1 or IDH2 are positive prognostic factors | [44] |
| ATRX | Prognostic | ATRX alterations are associated with a better prognosis | [32] |
| TERT | Diagnostic and prognostic | Mutations in the TERT gene are associated with a worse prognosis | [32] |
| CDKN2A | Prognostic | Homozygous deletion of this gene is associated with a poor prognosis | [32] |
| 1p/19q Codeletion | Prognostic | 1p/19q deletion is associated with a more favourable clinical outcome, although this alteration is uncommon in glioblastoma | [49] |
| 7+/10− | Diagnostic and prognostic | Gain of chromosome 7 and loss of chromosome 10 are common alterations in glioblastoma and are typically associated with a poor prognosis | [32] |
| EGFR | Diagnostic, prognostic and predictive | Amplification of this gene is associated with a poor prognosis and resistance to therapy | [52,54] |
| MGMT | Prognostic and predictive | Promoter methylation of the MGMT gene is associated with a favourable prognosis and a better response to treatment | [32] |
5.9. Liquid Biopsy
5.9.1. Circulating Tumour Cells
5.9.2. Cell-Free Nucleic Acids
5.9.3. Extracellular Vesicles
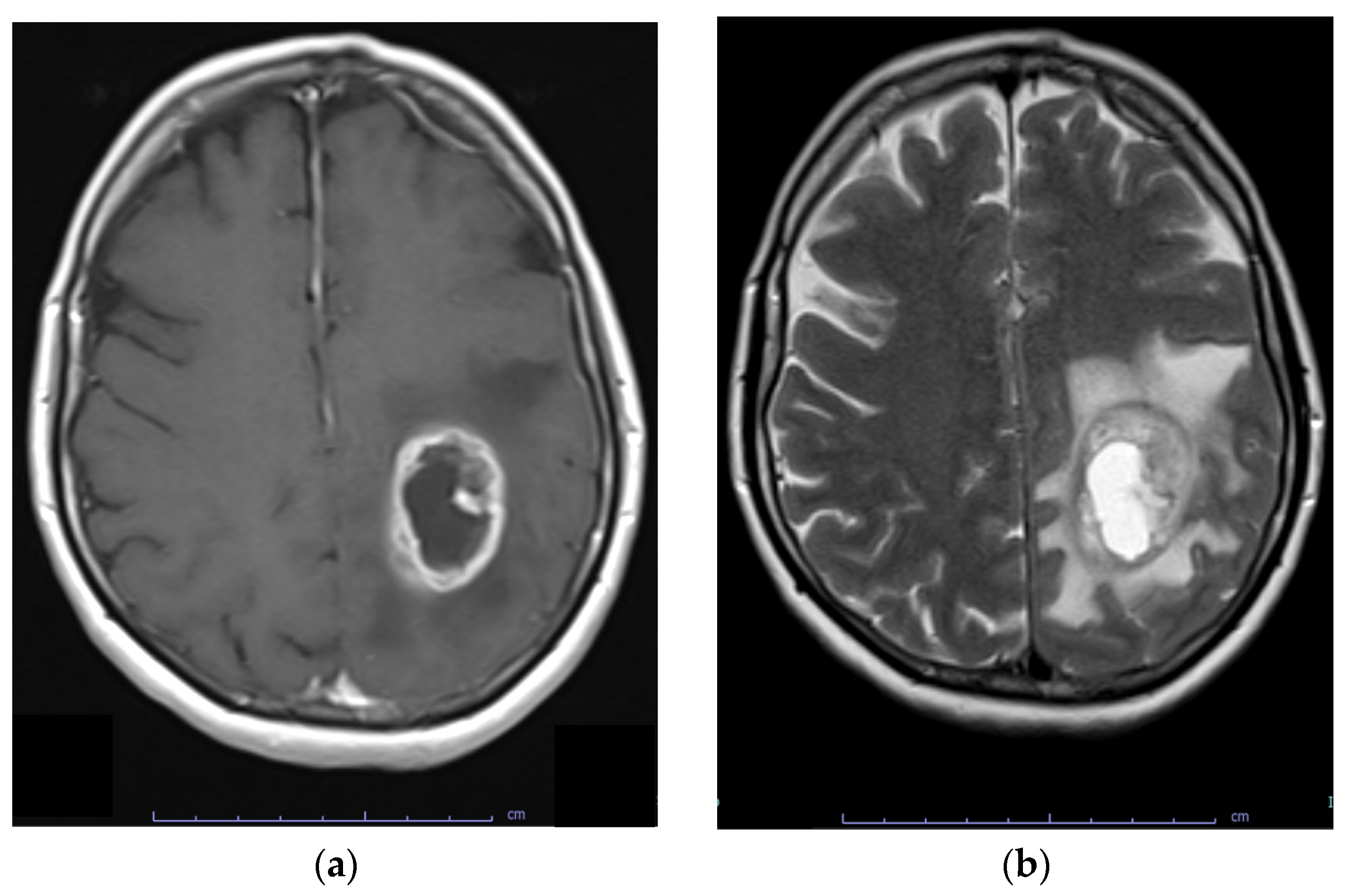
6. Diagnosis
7. Current Treatment Options
7.1. Management of Newly Diagnosed Glioblastoma
7.2. Radiotherapy Considerations
8. Novel Treatment Options
8.1. Targeted Therapies: From Cell Signalling Pathways to Cell Metabolism
8.2. Immunotherapies
8.3. Obstacles in Using Targeted Therapies
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perry, A.; Wesseling, P. Histologic Classification of Gliomas. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 71–95. [Google Scholar]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; Codon Publications: Singapore, 2017; pp. 143–153. [Google Scholar]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of Glioblastoma: State of the Art and Future Directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef]
- Sidaway, P. Glioblastoma Subtypes Revisited. Nat. Rev. Clin. Oncol. 2017, 14, 587. [Google Scholar] [CrossRef]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme–Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Patel, N.P.; Lyon, K.A.; Huang, J.H. The Effect of Race on the Prognosis of the Glioblastoma Patient: A Brief Review. Neurol. Res. 2019, 41, 967–971. [Google Scholar] [CrossRef]
- Kyritsis, A.P.; Bondy, M.L.; Rao, J.S.; Sioka, C. Inherited Predisposition to Glioma. Neuro-Oncology 2010, 12, 104–113. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of Its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Vienne-Jumeau, A.; Tafani, C.; Ricard, D. Environmental Risk Factors of Primary Brain Tumors: A Review. Rev. Neurol. 2019, 175, 664–678. [Google Scholar] [CrossRef]
- Rasheed, S.; Rehman, K.; Akash, M.S.H. An Insight into the Risk Factors of Brain Tumors and Their Therapeutic Interventions. Biomed. Pharmacother. 2021, 143, 112119. [Google Scholar] [CrossRef]
- Gatto, N.M.; Ogata, P.; Lytle, B. Farming, Pesticides, and Brain Cancer: A 20-Year Updated Systematic Literature Review and Meta-Analysis. Cancers 2021, 13, 4477. [Google Scholar] [CrossRef]
- Bielecka, J.; Markiewicz-żukowska, R. The Influence of Nutritional and Lifestyle Factors on Glioma Incidence. Nutrients 2020, 12, 1812. [Google Scholar] [CrossRef]
- Schüz, J.; Pirie, K.; Reeves, G.K.; Floud, S.; Beral, V. Cellular Telephone Use and the Risk of Brain Tumors: Update of the UK Million Women Study. J. Natl. Cancer Inst. 2022, 114, 704–711. [Google Scholar] [CrossRef]
- Inskip, P.D.; Tarone, R.E.; Hatch, E.E.; Wilcosky, T.C.; Shapiro, W.R.; Selker, R.G.; Fine, H.A.; Black, P.M.; Loeffler, J.S.; Linet, M.S. Cellular-Telephone Use and Brain Tumors. N. Engl. J. Med. 2001, 344, 79–86. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Sejda, A.; Grajkowska, W.; Trubicka, J.; Szutowicz, E.; Wojdacz, T.; Kloc, W.; Iżycka-Świeszewska, E. WHO CNS5 2021 Classification of Gliomas: A Practical Review and Road Signs for Diagnosing Pathologists and Proper Patho-Clinical and Neuro-Oncological Cooperation. Folia Neuropathol. 2022, 60, 137–152. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Burger, P.; Ellison, D.W.; Reifenberger, G.; von Deimling, A.; Aldape, K.; Brat, D.; Collins, V.P.; Eberhart, C.; et al. International Society of Neuropathology—Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014, 24, 429–435. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Torp, S.H.; Solheim, O.; Skjulsvik, A.J. The WHO 2021 Classification of Central Nervous System Tumours: A Practical Update on What Neurosurgeons Need to Know—A Minireview. Acta Neurochir. 2022, 164, 2453–2464. [Google Scholar] [CrossRef]
- Nabors, B.; Portnow, J.; Hattangadi-Gluth, J.; Horbinski, C. NCCN CNS Tumor Guidelines Update for 2023. Neuro-Oncology 2023, 25, 2114–2116. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Esemen, Y.; Awan, M.; Parwez, R.; Baig, A.; Rahman, S.; Masala, I.; Franchini, S.; Giakoumettis, D. Molecular Pathogenesis of Glioblastoma in Adults and Future Perspectives: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 2607. [Google Scholar] [CrossRef]
- Mao, H.; LeBrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated Signaling Pathways in Glioblastoma Multiforme: Molecular Mechanisms and Therapeutic Targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef]
- Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers 2021, 13, 5059. [Google Scholar] [CrossRef]
- El Atat, O.; Naser, R.; Abdelkhalek, M.; Habib, R.; El Sibai, M. Molecular Targeted Therapy: A New Avenue in Glioblastoma Treatment (Review). Oncol. Lett. 2022, 25, 46. [Google Scholar] [CrossRef]
- Di Nunno, V.; Gatto, L.; Tosoni, A.; Bartolini, S.; Franceschi, E. Implications of BRAF V600E Mutation in Gliomas: Molecular Considerations, Prognostic Value and Treatment Evolution. Front. Oncol. 2023, 12, 1067252. [Google Scholar] [CrossRef]
- Pearson, J.R.D.; Regad, T. Targeting Cellular Pathways in Glioblastoma Multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Mamun, A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of Glioblastoma Multiforme: From Molecular Mechanisms to Therapeutic Approaches. Semin. Cancer Biol. 2022, 83, 100–120. [Google Scholar] [CrossRef]
- Romani, M.; Pistillo, M.P.; Banelli, B. Epigenetic Targeting of Glioblastoma. Front. Oncol. 2018, 8, 448. [Google Scholar] [CrossRef]
- Śledzińska, P.; Bebyn, M.G.; Furtak, J.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Biomarkers in Gliomas. Int. J. Mol. Sci. 2021, 22, 10373. [Google Scholar] [CrossRef]
- Chen, H.-M.; Nikolic, A.; Singhal, D.; Gallo, M. Roles of Chromatin Remodelling and Molecular Heterogeneity in Therapy Resistance in Glioblastoma. Cancers 2022, 14, 4942. [Google Scholar] [CrossRef]
- Ganguly, D.; Sims, M.; Cai, C.; Fan, M.; Pfeffer, L.M. Chromatin Remodeling Factor BRG1 Regulates Stemness and Chemosensitivity of Glioma Initiating Cells. Stem Cells 2018, 36, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Goenka, A.; Tiek, D.M.; Song, X.; Iglesia, R.P.; Lu, M.; Hu, B.; Cheng, S.-Y. The Role of Non-Coding RNAs in Glioma. Biomedicines 2022, 10, 2031. [Google Scholar] [CrossRef]
- McClellan, B.L.; Haase, S.; Nunez, F.J.; Alghamri, M.S.; Dabaja, A.A.; Lowenstein, P.R.; Castro, M.G. Impact of Epigenetic Reprogramming on Antitumor Immune Responses in Glioma. J. Clin. Investig. 2023, 133, e163450. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R.P.; Costello, J.F. Epigenetic Mechanisms in Glioblastoma Multiforme. Semin. Cancer Biol. 2009, 19, 188–197. [Google Scholar] [CrossRef]
- Wu, Q.; Berglund, A.E.; Etame, A.B. The Impact of Epigenetic Modifications on Adaptive Resistance Evolution in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8324. [Google Scholar] [CrossRef]
- Do, H.; Kim, W. Roles of Oncogenic Long Non-Coding RNAs in Cancer Development. Genom. Inform. 2018, 16, e18. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Pal, S.; Rubstov, Y.; Goel, A.; Garg, M.; Pavlyukov, M.; Pandey, A.K. LncRNAs Associated with Glioblastoma: From Transcriptional Noise to Novel Regulators with a Promising Role in Therapeutics. Mol. Ther. Nucleic Acids 2021, 24, 728–742. [Google Scholar] [CrossRef]
- Behrooz, A.B.; Latifi-Navid, H.; da Silva Rosa, S.C.; Swiat, M.; Wiechec, E.; Vitorino, C.; Vitorino, R.; Jamalpoor, Z.; Ghavami, S. Integrating Multi-Omics Analysis for Enhanced Diagnosis and Treatment of Glioblastoma: A Comprehensive Data-Driven Approach. Cancers 2023, 15, 3158. [Google Scholar] [CrossRef]
- Rana, R.; Kumari, B.; Kumari, J.; Ganguly, N.K. Glioblastoma Diagnostics and Prognostic Biomarkers: Current Status in Medicine and Exosome Derivation. Curr. Med. Res. Pract. 2019, 9, 65–73. [Google Scholar] [CrossRef]
- Sharma, N.; Mallela, A.N.; Shi, D.D.; Tang, L.W.; Abou-Al-Shaar, H.; Gersey, Z.C.; Zhang, X.; McBrayer, S.K.; Abdullah, K.G. Isocitrate Dehydrogenase Mutations in Gliomas: A Review of Current Understanding and Trials. Neurooncol. Adv. 2023, 5, vdad053. [Google Scholar] [CrossRef] [PubMed]
- Kayabolen, A.; Yilmaz, E.; Bagci-Onder, T. IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications? Biomedicines 2021, 9, 799. [Google Scholar] [CrossRef] [PubMed]
- Hasanau, T.; Pisarev, E.; Kisil, O.; Nonoguchi, N.; Le Calvez-Kelm, F.; Zvereva, M. Detection of TERT Promoter Mutations as a Prognostic Biomarker in Gliomas: Methodology, Prospects, and Advances. Biomedicines 2022, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- Arita, H.; Narita, Y.; Fukushima, S.; Tateishi, K.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Collins, V.P.; Kawahara, N.; et al. Upregulating Mutations in the TERT Promoter Commonly Occur in Adult Malignant Gliomas and Are Strongly Associated with Total 1p19q Loss. Acta Neuropathol. 2013, 126, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Guo, Y.; Zhao, D.; Zou, Q.; Yu, F.; Zhang, L.; Xu, L. Comprehensive Analysis Revealed That CDKN2A Is a Biomarker for Immune Infiltrates in Multiple Cancers. Front. Cell Dev. Biol. 2021, 9, 808208. [Google Scholar] [CrossRef]
- Clark, K.H.; Villano, J.L.; Nikiforova, M.N.; Hamilton, R.L.; Horbinski, C. 1p/19q Testing Has No Significance in the Workup of Glioblastomas. Neuropathol. Appl. Neurobiol. 2013, 39, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, M.; Yoshimoto, K.; Ma, X.; Guan, Y.; Hata, N.; Amano, T.; Nakamizo, A.; Suzuki, S.O.; Iwaki, T.; Sasaki, T. Molecular Characteristics of Glioblastoma with 1p/19q Co-Deletion. Brain Tumor Pathol. 2012, 29, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hänggi, D.; Wick, W.; et al. Distribution of EGFR Amplification, Combined Chromosome 7 Gain and Chromosome 10 Loss, and TERT Promoter Mutation in Brain Tumors and Their Potential for the Reclassification of IDHwt Astrocytoma to Glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Benitez, J.A.; Ma, J.; D’Antonio, M.; Boyer, A.; Camargo, M.F.; Zanca, C.; Kelly, S.; Khodadadi-Jamayran, A.; Jameson, N.M.; Andersen, M.; et al. PTEN Regulates Glioblastoma Oncogenesis through Chromatin-Associated Complexes of DAXX and Histone H3.3. Nat. Commun. 2017, 8, 15223. [Google Scholar] [CrossRef]
- Senhaji, N.; Houssaini, A.S.; Lamrabet, S.; Louati, S.; Bennis, S. Molecular and Circulating Biomarkers in Patients with Glioblastoma. Int. J. Mol. Sci. 2022, 23, 7474. [Google Scholar] [CrossRef]
- Ludwig, K.; Kornblum, H.I. Molecular Markers in Glioma. J. Neurooncol. 2017, 134, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII Variant in Glioblastoma Multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.-I.; et al. Prognostic Value of Epidermal Growth Factor Receptor in Patients with Glioblastoma Multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar] [PubMed]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic Significance of Epidermal Growth Factor Receptor Expression in Glioma Patients. OncoTargets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, K.; Das, B.; Singh, A.K.; Misra, A.; Misra, S.; Misra, S.S. Prognostic Significance of Epidermal Growth Factor Receptor in Patients of Glioblastoma Multiforme. J. Clin. Diagn. Res. 2017, 11, EC05. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic Pathways to Glioblastoma. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, C.; Palmer, A.; Williams, H.; Wragg, C.; Haynes, H.R.; White, P.; DeSouza, R.-M.; Williams, M.; Hopkins, K.; Kurian, K.M. EGFR and EGFRvIII Analysis in Glioblastoma as Therapeutic Biomarkers. Br. J. Neurosurg. 2015, 29, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Pongor, L.; Su, Y.-T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhang, W.; Wang, Y.; Peng, X.; Chen, B.; Qiu, X.; Li, G.; Li, S.; Wu, C.; Yao, K.; et al. IDH Mutation and MGMT Promoter Methylation in Glioblastoma: Results of a Prospective Registry. Oncotarget 2015, 6, 40896–40906. [Google Scholar] [CrossRef]
- Radke, J.; Koch, A.; Pritsch, F.; Schumann, E.; Misch, M.; Hempt, C.; Lenz, K.; Löbel, F.; Paschereit, F.; Heppner, F.L.; et al. Predictive MGMT Status in a Homogeneous Cohort of IDH Wildtype Glioblastoma Patients. Acta Neuropathol. Commun. 2019, 7, 89. [Google Scholar] [CrossRef]
- Annavarapu, S.; Gogate, A.; Pham, T.; Davies, K.; Singh, P.; Robert, N. Treatment Patterns and Outcomes for Patients with Newly Diagnosed Glioblastoma Multiforme: A Retrospective Cohort Study. CNS Oncol. 2021, 10, CNS76. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.; Li, G.; Liu, Y.; Zhang, K.; Zhao, Z.; Wu, F.; Chang, Y.; Pang, B.; Li, J.; Li, Y.; et al. Predictive Value of MGMT Promoter Methylation on the Survival of TMZ Treated IDH-Mutant Glioblastoma. Cancer Biol. Med. 2021, 18, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Szylberg, M.; Sokal, P.; Śledzińska, P.; Bebyn, M.; Krajewski, S.; Szylberg, Ł.; Szylberg, A.; Szylberg, T.; Krystkiewicz, K.; Birski, M.; et al. MGMT Promoter Methylation as a Prognostic Factor in Primary Glioblastoma: A Single-Institution Observational Study. Biomedicines 2022, 10, 2030. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O6-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2019, 9, 1547. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal Fluid-Derived Circulating Tumour DNA Better Represents the Genomic Alterations of Brain Tumours than Plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J.; Till, J.; Abdalla, A.; Sangha, H.K.; Yee, S.S.; Freedman, J.; Black, T.A.; Hussain, J.; Binder, Z.A.; Brem, S.; et al. Association of Plasma Cell-Free DNA with Survival in Patients with IDH Wild-Type Glioblastoma. Neurooncol. Adv. 2021, 3, vdab011. [Google Scholar] [CrossRef]
- Mouliere, F.; Smith, C.G.; Heider, K.; Su, J.; van der Pol, Y.; Thompson, M.; Morris, J.; Wan, J.C.M.; Chandrananda, D.; Hadfield, J.; et al. Fragmentation Patterns and Personalized Sequencing of Cell-free DNA in Urine and Plasma of Glioma Patients. EMBO Mol. Med. 2021, 13, e12881. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma Heterogeneity at Single Cell Resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Eibl, R.H.; Schneemann, M. Liquid Biopsy and Glioblastoma. Explor. Target. Antitumor Ther. 2023, 4, 28–41. [Google Scholar] [CrossRef]
- Pentsova, E.I.; Shah, R.H.; Tang, J.; Boire, A.; You, D.; Briggs, S.; Omuro, A.; Lin, X.; Fleisher, M.; Grommes, C.; et al. Evaluating Cancer of the Central Nervous System through Next-Generation Sequencing of Cerebrospinal Fluid. J. Clin. Oncol. 2016, 34, 2404–2415. [Google Scholar] [CrossRef]
- Martínez-Ricarte, F.; Mayor, R.; Martínez-Sáez, E.; Rubio-Pérez, C.; Pineda, E.; Cordero, E.; Cicuéndez, M.; Poca, M.A.; López-Bigas, N.; Ramon y Cajal, S.; et al. Molecular Diagnosis of Diffuse Gliomas through Sequencing of Cell-Free Circulating Tumor DNA from Cerebrospinal Fluid. Clin. Cancer Res. 2018, 24, 2812–2819. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Diplas, B.H.; Chen, X.; Wu, Y.; Xiao, X.; Jiang, L.; Geng, Y.; Xu, C.; Sun, Y.; Zhang, P.; et al. Molecular Profiling of Tumors of the Brainstem by Sequencing of CSF-Derived Circulating Tumor DNA. Acta Neuropathol. 2019, 137, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Hickman, R.A.; Miller, A.M.; Arcila, M.E. Cerebrospinal Fluid: A Unique Source of Circulating Tumor DNA with Broad Clinical Applications. Transl. Oncol. 2023, 33, 101688. [Google Scholar] [CrossRef] [PubMed]
- Bark, J.M.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating Biomarkers in Patients with Glioblastoma. Br. J. Cancer 2020, 122, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Pellerino, A.; Soffietti, R.; Rudà, R. Blood–Brain Barrier in Brain Tumors: Biology and Clinical Relevance. Int. J. Mol. Sci. 2021, 22, 12654. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Turner, S.; Peters, K.B.; Desjardins, A.; Gururangan, S.; Sampson, J.H.; McLendon, R.E.; Herndon, J.E.; Jones, L.W.; Kirkpatrick, J.P.; et al. A Review of VEGF/VEGFR-Targeted Therapeutics for Recurrent Glioblastoma. J. Natl. Compr. Cancer Netw. 2011, 9, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Husain, H.; Fanta, P.T.; Piccioni, D.E.; Kesari, S.; Schwab, R.B.; Banks, K.C.; Lanman, R.B.; Talasaz, A.; Parker, B.A.; et al. Detection Rate of Actionable Mutations in Diverse Cancers Using a Biopsy-Free (Blood) Circulating Tumor Cell DNA Assay. Oncotarget 2016, 7, 9707–9717. [Google Scholar] [CrossRef]
- Piccioni, D.E.; Achrol, A.S.; Kiedrowski, L.A.; Banks, K.C.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of Cell-Free Circulating Tumor DNA in 419 Patients with Glioblastoma and Other Primary Brain Tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef]
- Bagley, S.J.; Nabavizadeh, S.A.; Mays, J.J.; Till, J.E.; Ware, J.B.; Levy, S.; Sarchiapone, W.; Hussain, J.; Prior, T.; Guiry, S.; et al. Clinical Utility of Plasma Cell-Free DNA in Adult Patients with Newly Diagnosed Glioblastoma: A Pilot Prospective Study. Clin. Cancer Res. 2020, 26, 397–407. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Oshi, M.; Murthy, V.; Takahashi, H.; Huyser, M.; Okano, M.; Tokumaru, Y.; Rashid, O.M.; Matsuyama, R.; Endo, I.; Takabe, K. Urine as a Source of Liquid Biopsy for Cancer. Cancers 2021, 13, 2652. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, T.R. A Case of Cancer in Which Cells Similar to Those in the Tumours Were Seen in the Blood after Death. Aus. Med. J. 1869, 14, 146–149. [Google Scholar]
- Alba-Bernal, A.; Lavado-Valenzuela, R.; Domínguez-Recio, M.E.; Jiménez-Rodriguez, B.; Queipo-Ortuño, M.I.; Alba, E.; Comino-Méndez, I. Challenges and Achievements of Liquid Biopsy Technologies Employed in Early Breast Cancer. EBioMedicine 2020, 62, 103100. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating Tumor Cells: Biology and Clinical Significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef] [PubMed]
- Lessi, F.; Morelli, M.; Franceschi, S.; Aretini, P.; Menicagli, M.; Marranci, A.; Pasqualetti, F.; Gambacciani, C.; Pieri, F.; Grimod, G.; et al. Innovative Approach to Isolate and Characterize Glioblastoma Circulating Tumor Cells and Correlation with Tumor Mutational Status. Int. J. Mol. Sci. 2023, 24, 10147. [Google Scholar] [CrossRef] [PubMed]
- Krol, I.; Castro-Giner, F.; Maurer, M.; Gkountela, S.; Szczerba, B.M.; Scherrer, R.; Coleman, N.; Carreira, S.; Bachmann, F.; Anderson, S.; et al. Detection of Circulating Tumour Cell Clusters in Human Glioblastoma. Br. J. Cancer 2018, 119, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.; Powter, B.; Po, J.W.; Cooper, A.; Garrett, C.; Koh, E.-S.; Sheridan, M.; van Gelder, J.; Darwish, B.; Mckechnie, S.; et al. Isolation of Circulating Tumor Cells from Glioblastoma Patients by Direct Immunomagnetic Targeting. Appl. Sci. 2020, 10, 3338. [Google Scholar] [CrossRef]
- Qi, Y.; Sun, Q.; Deng, G.; Zhang, H.; Xu, Y.; Li, Y.; Huang, S.; Li, Y.; Ye, Z.; Wang, Y.; et al. Identifying Circulating Glioma Cells and Their Clusters as Diagnostic Markers by a Novel Detection Platform. Clin. Transl. Med. 2021, 11, e318. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Holtschmidt, J.; Auer, M.; Heitzer, E.; Lamszus, K.; Schulte, A.; Matschke, J.; Langer-Freitag, S.; Gasch, C.; Stoupiec, M.; et al. Hematogenous Dissemination of Glioblastoma Multiforme. Sci. Transl. Med. 2014, 6, 247ra101. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain Tumor Cells in Circulation Are Enriched for Mesenchymal Gene Expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef]
- Gatto, L.; Franceschi, E.; Di Nunno, V.; Tosoni, A.; Lodi, R.; Brandes, A.A. Liquid Biopsy in Glioblastoma Management: From Current Research to Future Perspectives. Oncologist 2021, 26, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Antoñanzas, A.; Auzmendi-Iriarte, J.; Carrasco-Garcia, E.; Moreno-Cugnon, L.; Ruiz, I.; Villanua, J.; Egaña, L.; Otaegui, D.; Samprón, N.; Matheu, A. Liquid Biopsy in Glioblastoma: Opportunities, Applications and Challenges. Cancers 2019, 11, 950. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Mishra, S.; Hadi, R.; Sahu, A.; Kumari, S.; Rastogi, M.; Khurana, R.; Shukla, S.; Siddiqui, M.H.; Husain, N. Dynamics of Cell-Free DNA in Predicting Response in Adult Diffuse Glioma on Chemoradiotherapy. Cancer Genet. 2022, 268–269, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Fontanilles, M.; Marguet, F.; Beaussire, L.; Magne, N.; Pépin, L.-F.; Alexandru, C.; Tennevet, I.; Hanzen, C.; Langlois, O.; Jardin, F.; et al. Cell-Free DNA and Circulating TERT Promoter Mutation for Disease Monitoring in Newly-Diagnosed Glioblastoma. Acta Neuropathol. Commun. 2020, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, K.; Yekula, A.; Small, J.L.; Rosh, Z.S.; Kang, K.M.; Wang, L.; Lau, S.; Zhang, H.; Lee, H.; Bettegowda, C.; et al. TERT Promoter Mutation Analysis for Blood-Based Diagnosis and Monitoring of Gliomas. Clin. Cancer Res. 2021, 27, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Liu, Z.; Zhu, Y.; Ma, L. Genome-Wide Methylation Analysis of Circulating Tumor DNA: A New Biomarker for Recurrent Glioblastom. Heliyon 2023, 9, e14339. [Google Scholar] [CrossRef] [PubMed]
- Zanganeh, S.; Abbasgholinejad, E.; Doroudian, M.; Esmaelizad, N.; Farjadian, F.; Benhabbour, S.R. The Current Landscape of Glioblastoma Biomarkers in Body Fluids. Cancers 2023, 15, 3804. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Medarova, Z.; Moore, A. Role of MicroRNAs in Glioblastoma. Oncotarget 2021, 12, 1707–1723. [Google Scholar] [CrossRef]
- Makowska, M.; Smolarz, B.; Romanowicz, H. MicroRNAs (MiRNAs) in Glioblastoma Multiforme (GBM)—Recent Literature Review. Int. J. Mol. Sci. 2023, 24, 3521. [Google Scholar] [CrossRef]
- Khristov, V.; Lin, A.; Freedman, Z.; Staub, J.; Shenoy, G.; Mrowczynski, O.; Rizk, E.; Zacharia, B.; Connor, J. Tumor-Derived Biomarkers in Liquid Biopsy of Glioblastoma. World Neurosurg. 2023, 170, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Su, J.; Long, W.; Qin, C.; Wang, X.; Xiao, K.; Li, Y.; Xiao, Q.; Wang, J.; Pan, Y.; et al. LINC00470 Promotes Tumour Proliferation and Invasion, and Attenuates Chemosensitivity through the LINC00470/MiR-134/Myc/ABCC1 Axis in Glioma. J. Cell. Mol. Med. 2020, 24, 12094–12106. [Google Scholar] [CrossRef] [PubMed]
- Chargaff, E.; West, R. The Biological Significance of the Thromboplastic Protein of Blood. J. Biol. Chem. 1946, 166, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Irmer, B.; Chandrabalan, S.; Maas, L.; Bleckmann, A.; Menck, K. Extracellular Vesicles in Liquid Biopsies as Biomarkers for Solid Tumors. Cancers 2023, 15, 1307. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, M.; Osti, D.; Faletti, S.; Beznoussenko, G.V.; DiMeco, F.; Pelicci, G. Extracellular Vesicles: The Key for Precision Medicine in Glioblastoma. Neuro-Oncology 2022, 24, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Nguyen, H.P.T.; Jones, J.J.; Stylli, S.S.; Whitehead, C.A.; Paradiso, L.; Luwor, R.B.; Areeb, Z.; Hanssen, E.; Cho, E.; et al. Extracellular Vesicles Secreted by Glioma Stem Cells Are Involved in Radiation Resistance and Glioma Progression. Int. J. Mol. Sci. 2022, 23, 2770. [Google Scholar] [CrossRef] [PubMed]
- Tzaridis, T.; Weller, J.; Bachurski, D.; Shakeri, F.; Schaub, C.; Hau, P.; Buness, A.; Schlegel, U.; Steinbach, J.; Seidel, C.; et al. A Novel Serum Extracellular Vesicle Protein Signature to Monitor Glioblastoma Tumor Progression. Int. J. Cancer 2023, 152, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.M.C.; Peterson, T.E.; Cepeda, M.A.; Johnson, A.J.; Parney, I.F. Isolation and Analysis of Plasma-Derived Exosomes in Patients with Glioma. Front. Oncol. 2019, 9, 651. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) Guideline on the Diagnosis and Treatment of Adult Astrocytic and Oligodendroglial Gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef]
- Wirsching, H.-G.; Galanis, E.; Weller, M. Glioblastoma. In Malignant Brain Tumors; Springer: Berlin/Heidelberg, Germany, 2016; pp. 381–397. [Google Scholar]
- Ly, K.I.; Wen, P.Y.; Huang, R.Y. Imaging of Central Nervous System Tumors Based on the 2016 World Health Organization Classification. Neurol. Clin. 2020, 38, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Capper, D.; Jeibmann, A.; Habel, A.; Paulus, W.; Dirk, T.; von Deimling, A. Addressing Diffuse Glioma as a Systemic Brain Disease with Single-Cell Analysis. Arch. Neurol. 2012, 69, 523. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.M.; Recinos, P.F.; Nowacki, A.S.; Schroeder, J.L.; Angelov, L.; Barnett, G.H.; Vogelbaum, M.A. Residual Tumor Volume Versus Extent of Resection: Predictors of Survival after Surgery for Glioblastoma. J. Neurosurg. 2014, 121, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Kreth, F.-W.; Thon, N.; Simon, M.; Westphal, M.; Schackert, G.; Nikkhah, G.; Hentschel, B.; Reifenberger, G.; Pietsch, T.; Weller, M.; et al. Gross Total but Not Incomplete Resection of Glioblastoma Prolongs Survival in the Era of Radiochemotherapy. Ann. Oncol. 2013, 24, 3117–3123. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Suchorska, B.; Weller, M.; Tabatabai, G.; Senft, C.; Hau, P.; Sabel, M.C.; Herrlinger, U.; Ketter, R.; Schlegel, U.; Marosi, C.; et al. Complete Resection of Contrast-Enhancing Tumor Volume Is Associated with Improved Survival in Recurrent Glioblastoma—Results from the DIRECTOR Trial. Neuro-Oncology 2016, 18, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A Multivariate Analysis of 416 Patients with Glioblastoma Multiforme: Prognosis, Extent of Resection, and Survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef]
- Shah, A.H.; Mahavadi, A.; Di, L.; Sanjurjo, A.; Eichberg, D.G.; Borowy, V.; Figueroa, J.; Luther, E.; de la Fuente, M.I.; Semonche, A.; et al. Survival Benefit of Lobectomy for Glioblastoma: Moving towards Radical Supramaximal Resection. J. Neuro-Oncol. 2020, 148, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.-J. Fluorescence-Guided Surgery with 5-Aminolevulinic Acid for Resection of Malignant Glioma: A Randomised Controlled Multicentre Phase III Trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Widhalm, G.; Stummer, W. What Is the Surgical Benefit of Utilizing 5-Aminolevulinic Acid for Fluorescence-Guided Surgery of Malignant Gliomas? Neurosurgery 2015, 77, 663–673. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Guzauskas, G.F.; Pollom, E.L.; Stieber, V.W.; Wang, B.C.M.; Garrison, L.P., Jr. Tumor Treating Fields and Maintenance Temozolomide for Newly-Diagnosed Glioblastoma: A Cost-Effectiveness Study. J. Med. Econ. 2019, 22, 1006–1013. [Google Scholar] [CrossRef]
- Keime-Guibert, F.; Chinot, O.; Taillandier, L.; Cartalat-Carel, S.; Frenay, M.; Kantor, G.; Guillamo, J.-S.; Jadaud, E.; Colin, P.; Bondiau, P.-Y.; et al. Radiotherapy for Glioblastoma in the Elderly. N. Engl. J. Med. 2007, 356, 1527–1535. [Google Scholar] [CrossRef]
- Kotecha, R.; Odia, Y.; Khosla, A.A.; Ahluwalia, M.S. Key Clinical Principles in the Management of Glioblastoma. JCO Oncol. Pract. 2023, 19, 180–189. [Google Scholar] [CrossRef]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef]
- Niyazi, M.; Andratschke, N.; Bendszus, M.; Chalmers, A.J.; Erridge, S.C.; Galldiks, N.; Lagerwaard, F.J.; Navarria, P.; af Rosenschöld, P.M.; Ricardi, U.; et al. ESTRO-EANO Guideline on Target Delineation and Radiotherapy Details for Glioblastoma. Radiother. Oncol. 2023, 184, 109663. [Google Scholar] [CrossRef] [PubMed]
- Niyazi, M.; Brada, M.; Chalmers, A.J.; Combs, S.E.; Erridge, S.C.; Fiorentino, A.; Grosu, A.L.; Lagerwaard, F.J.; Minniti, G.; Mirimanoff, R.-O.; et al. ESTRO-ACROP Guideline “Target Delineation of Glioblastomas”. Radiother. Oncol. 2016, 118, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, B.J.; Dobelbower, M.C.; Ennis, W.H.; Bag, A.K.; Markert, J.M.; Fiveash, J.B. Patterns of Failure for Glioblastoma Multiforme Following Limited-Margin Radiation and Concurrent Temozolomide. Radiat. Oncol. 2014, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Jette, D.; Chen, W. Creating a Spread-out Bragg Peak in Proton Beams. Phys. Med. Biol. 2011, 56, N131–N138. [Google Scholar] [CrossRef]
- Matsuda, M.; Mizumoto, M.; Kohzuki, H.; Sugii, N.; Sakurai, H.; Ishikawa, E. High-Dose Proton Beam Therapy ver-sus Conventional Fractionated Radiation Therapy for Newly Diagnosed Glioblastoma: A Propensity Score Match-ing Analysis. Radiat. Oncol. 2023, 18, 38. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma Targeted Therapy: Insight into Future of Molecular Approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients with Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; van den Bent, M.J.; Taphoorn, M.J.B.; Steuve, J.; Brandes, A.A.; Hamou, M.-F.; Wick, A.; et al. Phase II Study of Radiotherapy and Temsirolimus Versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [PubMed]
- Phuphanich, S.; Raizer, J.; Chamberlain, M.; Canelos, P.; Narwal, R.; Hong, S.; Miday, R.; Nade, M.; Laubscher, K. Phase II Study of MEDI-575, an Anti-Platelet-Derived Growth Factor-α Antibody, in Patients with Recurrent Glioblastoma. J. Neuro-Oncol. 2017, 131, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients with Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O6-Methylguanine–DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Sepúlveda-Sánchez, J.M.; Cloughesy, T.F.; Gil-Gil, M.J.; Puduvalli, V.K.; Raizer, J.J.; De Vos, F.Y.F.; Wen, P.Y.; Butowski, N.A.; Clement, P.M.J.; et al. Infigratinib in Patients with Recurrent Gliomas and FGFR Alterations: A Multicenter Phase II Study. Clin. Cancer Res. 2022, 28, 2270–2277. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.A.; Layer, J.P.; Leonardelli, S.; Friker, L.L.; Seidel, C.; Schaub, C.; Turiello, R.; Sperk, E.; Grau, F.; Paech, D.; et al. Radiotherapy and Olaptesed Pegol (NOX-A12) in Partially Resected or Biopsy-Only MGMT-Unmethylated Glioblastoma: Interim Data from the German Multicenter Phase 1/2 GLORIA Trial. J. Clin. Oncol. 2022, 40, 2050. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.E.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.J.; Buter, J.; Honkoop, A.H.; Boerman, D.; de Vos, F.Y.F.; et al. Single-Agent Bevacizumab or Lomustine Versus a Combination of Bevacizumab Plus Lomustine in Patients with Recurrent Glioblastoma (BELOB Trial): A Randomised Controlled Phase 2 Trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef]
- Lee, A.; Arasaratnam, M.; Chan, D.L.H.; Khasraw, M.; Howell, V.M.; Wheeler, H. Anti-Epidermal Growth Factor Receptor Therapy for Glioblastoma in Adults. Cochrane Database Syst. Rev. 2020, 5, CD013238. [Google Scholar] [CrossRef]
- Hernández, A.; Domènech, M.; Muñoz-Mármol, A.M.; Carrato, C.; Balana, C. Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment. Cells 2021, 10, 3529. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, C.; Reita, D.; Martin, S.; Entz-Werle, N.; Dontenwill, M. Glioblastoma Metabolism: Insights and Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 9137. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.; Duque, A.E.D.; Michalek, J.; Konkel, B.; Caflisch, L.; Chen, Y.; Pathuri, S.C.; Madhusudanannair-Kunnuparampil, V.; Floyd, J.; Brenner, A. Phase II Investigation of TVB-2640 (Denifanstat) with Bevacizumab in Patients with First Relapse High-Grade Astrocytoma. Clin. Cancer Res. 2023, 29, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Altwairgi, A.K.; Alghareeb, W.A.; AlNajjar, F.H.; Alhussain, H.; Alsaeed, E.; Balbaid, A.A.O.; Aldanan, S.; Orz, Y.; Alsharm, A.A. Atorvastatin in Combination with Radiotherapy and Temozolomide for Glioblastoma: A Prospective Phase II Study. Investig. New Drugs 2021, 39, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Sener, U.; Ruff, M.W.; Campian, J.L. Immunotherapy in Glioblastoma: Current Approaches and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 7046. [Google Scholar] [CrossRef] [PubMed]
- Bausart, M.; Préat, V.; Malfanti, A. Immunotherapy for Glioblastoma: The Promise of Combination Strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef] [PubMed]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma. JAMA Oncol. 2017, 3, 1094. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with Temozolomide for Patients with Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Jacques, F.H.; Nicholas, G.; Lorimer, I.A.J.; Foko, V.S.; Prevost, J.; Dumais, N.; Milne, K.; Nelson, B.H.; Woulfe, J.; Jansen, G.; et al. Avelumab in Newly Diagnosed Glioblastoma. Neurooncol. Adv. 2021, 3, vdab118. [Google Scholar] [CrossRef] [PubMed]
- Weathers, S.-P.S.; Kamiya-Matsuoka, C.; Harrison, R.A.; Liu, D.D.; Dervin, S.; Yun, C.; Loghin, M.E.; Penas-Prado, M.; Majd, N.; Yung, W.K.A.; et al. Phase I/II Study to Evaluate the Safety and Clinical Efficacy of Atezolizumab (Atezo; APDL1) in Combination with Temozolomide (TMZ) and Radiation in Patients with Newly Diagnosed Glioblastoma (GBM). J. Clin. Oncol. 2020, 38, 2511. [Google Scholar] [CrossRef]
- Weiss, T.; Hemmerle, T.; Roth, P.; Neri, D.; Weller, M. 378TiP A Phase I/II/IIb Study to Evaluate the Safety and Efficacy of the Tumor-Targeting Human Antibody-Cytokine Fusion Protein L19TNF Plus Standard Temozolomide Chemoradiotherapy in Patients with Newly Diagnosed Glioblastoma. Ann. Oncol. 2021, 32, S528. [Google Scholar] [CrossRef]





| Trial | Molecular Target | Therapy | Status | |
|---|---|---|---|---|
| Cell signalling pathways | NCT01339052 | PI3K | Buparlisib | Completed (phase II) |
| NCT00943826 | VEGF | Bevacizumab + TMZ | Completed (phase III) | |
| NCT01019434 | mTOR | Temsirolimus + RT | Completed (phase II) | |
| NCT05222802 | EGFR | ERAS-801 | Recruiting (phase I) | |
| NCT02981940 | CDK4/6 | Abemaciclib | Active, not recruiting (phase II) | |
| NCT01268566 | PDGFR | MEDI-575 | Completed (phase II) | |
| NCT01632228 | MET | Onartuzumab | Completed (phase II) | |
| NCT05376800 | MDM2 | Brigimadlin | Recruiting (phase 0/Ia) | |
| NCT01975701 | FGFR | BGJ398 | Completed (phase II) | |
| NCT02340156 | p53 | SGT-53 + TMZ | Terminated (phase II) | |
| NCT02345824 | CDK4/6 | Ribociclib | Unknown status (phase I) | |
| NCT04121455 | CXCL12 | NOX-A12 + RT | Active, not recruiting (phase I/II) | |
| NCT06102525 | hTERT | RZ-001 + Valganciclovir | Not yet recruiting (phase I/IIa) | |
| NCT01582269 | TGF-β | Galunisertib | Active, not recruiting (phase II) | |
| Tumour cell metabolism | NCT04825275 | HK2—glucose metabolism | Posaconazole | Recruiting (phase 0) |
| NCT04587830 | Arginine metabolism | ADI-PEG 20 + RT + TMZ | Recruiting (phase Ib) | |
| NCT03032484 | FASN—lipid metabolism | TVB-2640 + Bevacizumab | Completed (phase II) | |
| NCT04869449 | HK2—glucose metabolism | Ketoconazole | Recruiting (early phase I) | |
| NCT02029573 | HMGCR—cholesterol Metabolism | Atorvastatin + RT + TMZ | Completed (phase II) |
| Trial | Molecular Target | Type of Therapy | Status |
|---|---|---|---|
| NCT03726515 | EGFRvIII | CAR T + Pembrolizumab | Completed (phase I) |
| NCT00895180 | PDGFR | Monoclonal antibody | Completed (phase II) |
| NCT01454596 | EGFRvIII | CAR T | Completed (phase I/II) |
| NCT00128635 | DNA–histone H1 complex | Monoclonal antibody | All completed (phase I and phase II the NCT00004017) |
| NCT00004017 | |||
| NCT00509301 | |||
| NCT01498328 | EGFRvIII | Vaccine | Completed (phase II) |
| NCT01109095 | HER2 | CAR T | Completed (phase I) |
| NCT04214392 | CLTX | CAR T | Recruiting (phase I) |
| NCT03174197 | PD-1/PD-L1 | Monoclonal antibody with a checkpoint inhibitor function + RT + TMZ | Active, not recruiting (phase I/II) |
| NCT04443010 | EDB-FN | Cytokines + TMZ | Recruiting (phase I/II) |
| NCT03047473 | PD-1/PD-L1 | Monoclonal antibody with a checkpoint inhibitor function | Completed (phase II) |
| NCT02049489 | CD133 | Vaccine | Completed (phase I) |
| NCT05024175 | EGFRvIII | CAR-T | Not yet recruiting (phase I) |
| NCT04047706 | PD-1/PD-L1 | Monoclonal antibody with a checkpoint inhibitor function | Active, not recruiting (phase I) |
| NCT02017717 | PD-1/PD-L1 | Monoclonal antibody with a checkpoint inhibitor function | Active, not recruiting (phase III) |
| NCT04661384 | IL13Rα2 | CAR T | Recruiting (phase I) |
| NCT01480479 | EGFRvIII | Vaccine + TMZ | Completed (phase III) |
| NCT04003649 | IL13Rα2 | CAR T with checkpoint inhibition | Recruiting (phase I) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roda, D.; Veiga, P.; Melo, J.B.; Carreira, I.M.; Ribeiro, I.P. Principles in the Management of Glioblastoma. Genes 2024, 15, 501. https://doi.org/10.3390/genes15040501
Roda D, Veiga P, Melo JB, Carreira IM, Ribeiro IP. Principles in the Management of Glioblastoma. Genes. 2024; 15(4):501. https://doi.org/10.3390/genes15040501
Chicago/Turabian StyleRoda, Domingos, Pedro Veiga, Joana Barbosa Melo, Isabel Marques Carreira, and Ilda Patrícia Ribeiro. 2024. "Principles in the Management of Glioblastoma" Genes 15, no. 4: 501. https://doi.org/10.3390/genes15040501
APA StyleRoda, D., Veiga, P., Melo, J. B., Carreira, I. M., & Ribeiro, I. P. (2024). Principles in the Management of Glioblastoma. Genes, 15(4), 501. https://doi.org/10.3390/genes15040501