Molecular Mechanisms of DNA Replication Checkpoint Activation


Abstract
:1. Introduction
2. Principles of Eukaryotic DNA Replication
2.1. Pre-RC Formation

2.2. Pre-IC Formation
2.3. The Initiation Complex (IC)
2.4. Elongation and Termination
3. Cell Cycle Regulation of DNA Synthesis
3.1. The Licensing Factor Hypothesis
3.2. Regulation of DNA Synthesis by CDKs
3.3. G1 to S-Phase Transition
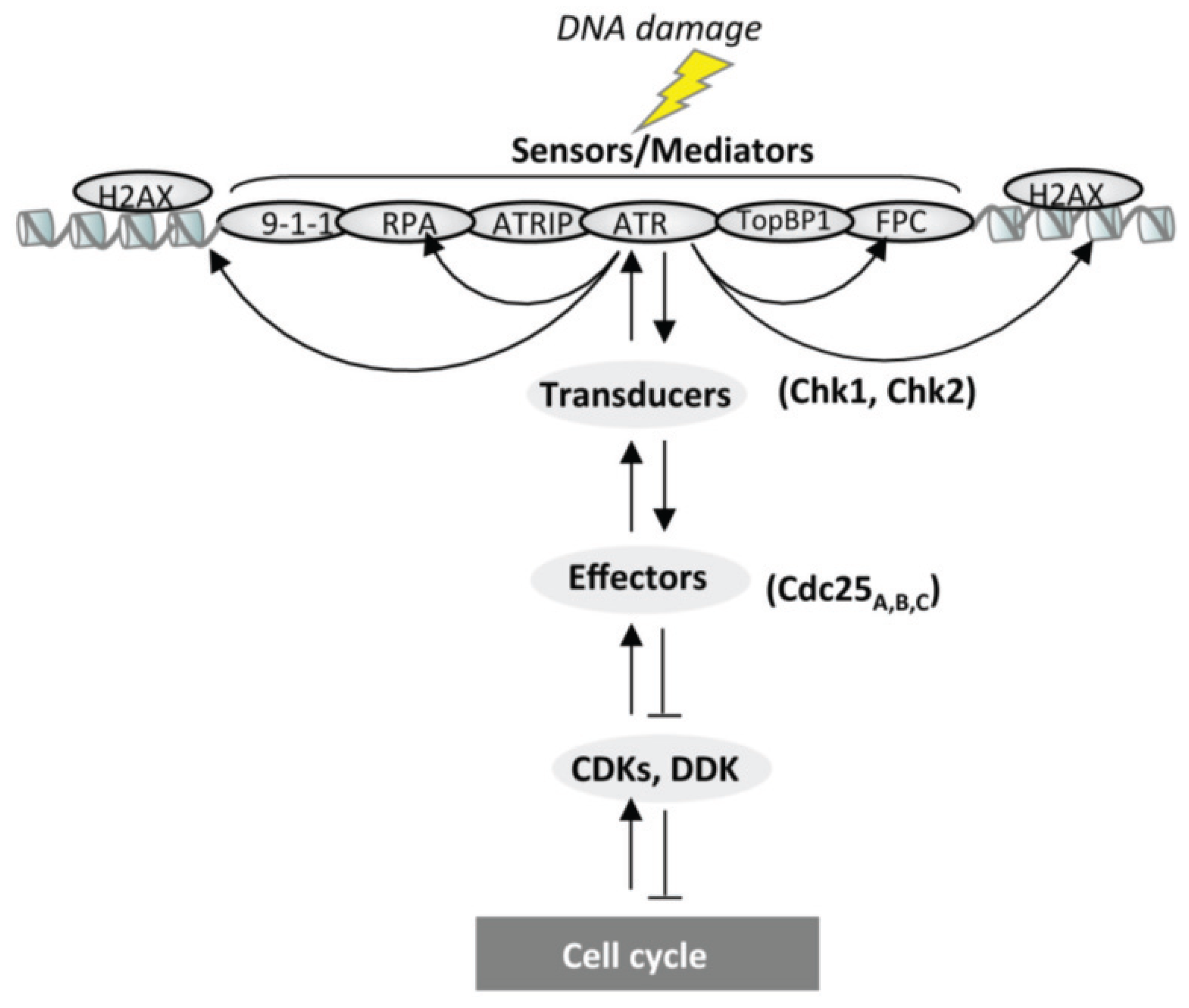
4. DNA Damage Response during S-Phase
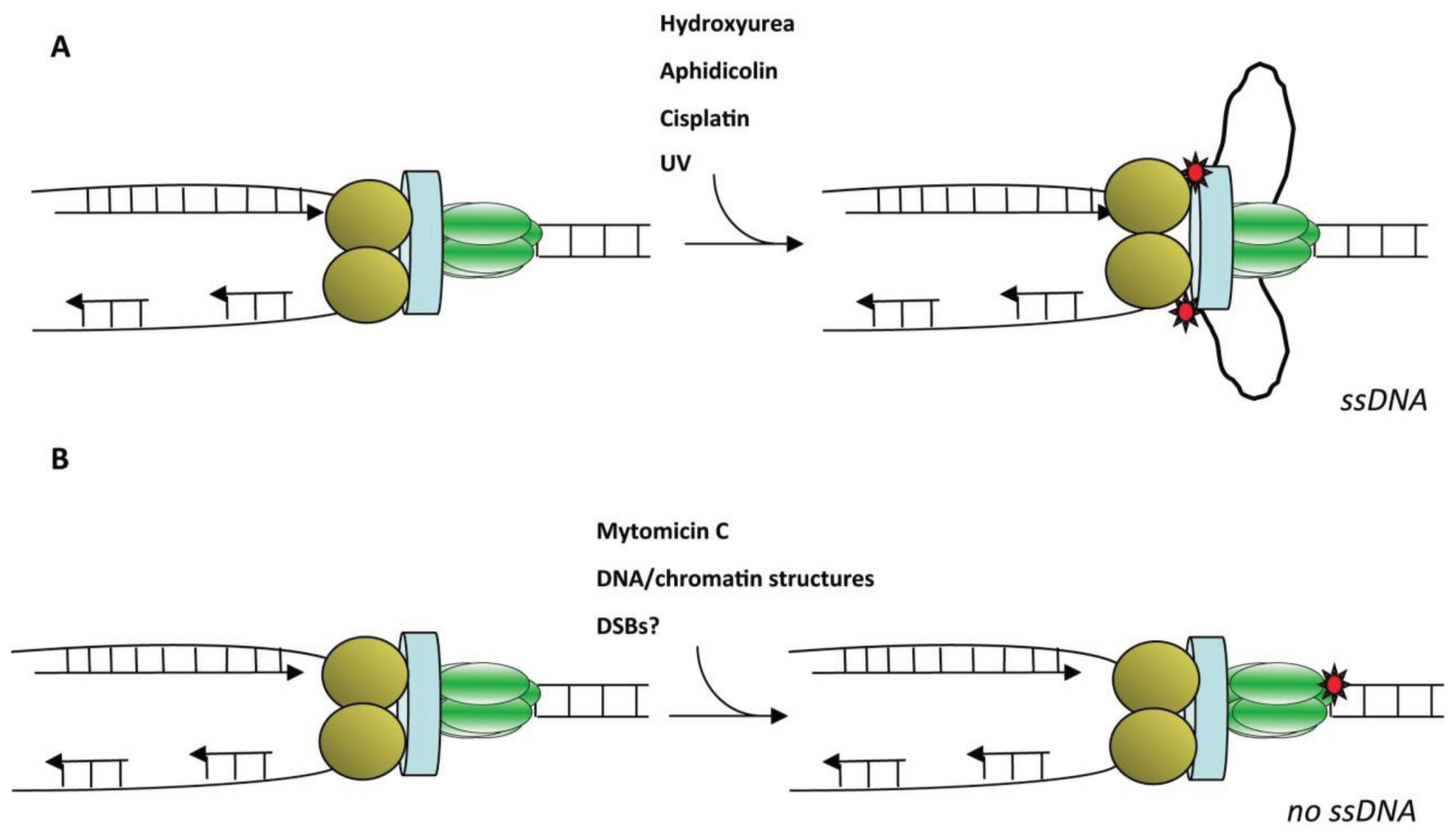
4.1. Sources of DNA Damage Acting as Obstacle for the Replisome
4.2. The Molecular Bases of the DNA Damage Response during S-Phase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Gene name | H. sapiens | X. laevis | D. melanogaster | C. elegans | Yeast(S. cerevisiae/S. pombe) |
|---|---|---|---|---|---|
| ATR | ATR | ATR | Mei-41 | atl-1 | Mec1 S.c/Rad3 S.p. |
| ATRIP | ATRIP | ATRIP | mus 304 | ATRIP | Ddc2/ATRIP |
| TopBP1 | TopBP1 | TopBP1 | mus 101 | mus 101 | Dbp11/Cut5 |
| Rad9 | Rad9 | Rad9 | Rad9 | Hrp-9 | Ddc1/Rad9 |
| Rad1 | Rad1 | Rad1 | Rad1/Dmel | mrt-2 | Rad17 |
| Hus1 | Hus1 | Hus1 | hus1 | hus-1 | Mec3/Hus1 |
| Rad17 | Rad17 | Rad17 | Rad17 | hrp-17 | Rad24/Rad17 |
| Claspin | Claspin | Claspin | Claspin | F25H5.5 | Mrc1/Mrc1 |
| Timeless | Timl | Tim 1 | Tim | tim-1 | Tof1/Swi1 |
| Tipin | Tipin | Tipin | Timeout | ? | Csm3/Swi3 |
| CHK1 | CHK1 | CHK1 | grp1/DChk1 | CHK1 | Chk1/Rad27 |
| CDC25A | CDC25A | CDC25A | String | CDC-25 | CDC25 |
5. The Replication Checkpoint
5.1. The S-Phase and Intra S-Phase Checkpoint
5.2. Generation of ssDNA after DNA Damage or Replicative Stress

5.3. Basic Model for ATR Activation at Stalled Replication Forks

5.4. Role of Replication Fork Uncoupling and RPA Nucleation onto ssDNA DNA Replication Checkpoint Activation
5.5. Cellular Responses to ATR Activation
Mediators and Transducers of the S-Phase Checkpoint
5.6. Transducers
5.7. Effector Proteins
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Donnell, M.; Langston, L.; Stillman, B. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb. Perspect. Biol. 2013. [Google Scholar] [CrossRef]
- Li, Y.; Araki, H. Loading and activation of DNA replicative helicases: The key step of initiation of DNA replication. Genes Cells 2013, 18, 266–277. [Google Scholar] [CrossRef]
- Symeonidou, I.E.; Taraviras, S.; Lygerou, Z. Control over DNA replication in time and space. FEBS Lett. 2012, 586, 2803–2812. [Google Scholar] [CrossRef]
- Bell, S.P.; Dutta, A. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 2002, 71, 333–374. [Google Scholar] [CrossRef]
- Remus, D.; Beuron, F.; Tolun, G.; Griffith, J.D.; Morris, E.P.; Diffley, J.F. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 2009, 139, 719–730. [Google Scholar] [CrossRef]
- Evrin, C.; Clarke, P.; Zech, J.; Lurz, R.; Sun, J.; Uhle, S.; Li, H.; Stillman, B.; Speck, C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 2009, 106, 20240–20245. [Google Scholar]
- Fernandez-Cid, A.; Riera, A.; Tognetti, S.; Herrera, M.C.; Samel, S.; Evrin, C.; Winkler, C.; Gardenal, E.; Uhle, S.; Speck, C. An ORC/Cdc6/MCM2-7 complex is formed in a multistep reaction to serve as a platform for MCM double-hexamer assembly. Mol. Cell 2013, 50, 577–588. [Google Scholar] [CrossRef]
- Frigola, J.; Remus, D.; Mehanna, A.; Diffley, J.F. ATPase-dependent quality control of DNA replication origin licensing. Nature 2013, 495, 339–343. [Google Scholar]
- McGarry, T.J.; Kirschner, M.W. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef]
- Tada, S.; Li, A.; Maiorano, D.; Mechali, M.; Blow, J.J. Repression of origin assembly in metaphase depends on inhibition of RLF-B/Cdt1 by geminin. Nat. Cell Biol. 2001, 3, 107–113. [Google Scholar] [CrossRef]
- Wohlschlegel, J.A.; Dwyer, B.T.; Dhar, S.K.; Cvetic, C.; Walter, J.C.; Dutta, A. Inhibition of eukaryotic DNA replication by geminin binding to cdt1. Science 2000, 290, 2309–2312. [Google Scholar] [CrossRef]
- Lutzmann, M.; Maiorano, D.; Mechali, M. Identification of full genes and proteins of MCM9, a novel, vertebrate-specific member of the MCM2-8 protein family. Gene 2005, 362, 51–56. [Google Scholar] [CrossRef]
- Lutzmann, M.; Mechali, M. MCM9 binds Cdt1 and is required for the assembly of prereplication complexes. Mol. Cell 2008, 31, 190–200. [Google Scholar] [CrossRef]
- Gambus, A.; Blow, J.J. Mcm8 and Mcm9 form a dimeric complex in Xenopus laevis egg extract that is not essential for DNA replication initiation. Cell Cycle 2013, 12, 1225–1232. [Google Scholar] [CrossRef]
- Lutzmann, M.; Grey, C.; Traver, S.; Ganier, O.; Maya-Mendoza, A.; Ranisavljevic, N.; Bernex, F.; Nishiyama, A.; Montel, N.; Gavois, E.; et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol. Cell 2012, 47, 523–534. [Google Scholar] [CrossRef]
- Park, J.; Long, D.T.; Lee, K.Y.; Abbas, T.; Shibata, E.; Negishi, M.; Luo, Y.; Schimenti, J.C.; Gambus, A.; Walter, J.C.; et al. The MCM8-MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Mol. Cell. Biol. 2013, 33, 1632–1644. [Google Scholar] [CrossRef]
- Nishimura, K.; Ishiai, M.; Horikawa, K.; Fukagawa, T.; Takata, M.; Takisawa, H.; Kanemaki, M.T. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol. Cell 2012, 47, 511–522. [Google Scholar] [CrossRef]
- Mendez, J.; Stillman, B. Perpetuating the double helix: Molecular machines at eukaryotic DNA replication origins. Bioessays 2003, 25, 1158–1167. [Google Scholar] [CrossRef]
- Mimura, S.; Takisawa, H. Xenopus Cdc45-dependent loading of DNA polymerase alpha onto chromatin under the control of S-phase Cdk. EMBO J. 1998, 17, 5699–5707. [Google Scholar] [CrossRef]
- Mimura, S.; Masuda, T.; Matsui, T.; Takisawa, H. Central role for cdc45 in establishing an initiation complex of DNA replication in Xenopus egg extracts. Genes Cells 2000, 5, 439–452. [Google Scholar] [CrossRef]
- Walter, J.; Newport, J. Initiation of eukaryotic DNA replication: Origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha. Mol. Cell 2000, 5, 617–627. [Google Scholar] [CrossRef]
- MacNeill, S.A. Structure and function of the GINS complex, a key component of the eukaryotic replisome. Biochem. J. 2010, 425, 489–500. [Google Scholar] [CrossRef]
- Costa, A.; Ilves, I.; Tamberg, N.; Petojevic, T.; Nogales, E.; Botchan, M.R.; Berger, J.M. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 2010, 18, 471–477. [Google Scholar]
- Sheu, Y.J.; Stillman, B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 2010, 463, 113–117. [Google Scholar] [CrossRef]
- Randell, J.C.; Fan, A.; Chan, C.; Francis, L.I.; Heller, R.C.; Galani, K.; Bell, S.P. Mec1 is one of multiple kinases that prime the Mcm2-7 helicase for phosphorylation by Cdc7. Mol. Cell 2010, 40, 353–363. [Google Scholar] [CrossRef]
- Tanaka, S.; Umemori, T.; Hirai, K.; Muramatsu, S.; Kamimura, Y.; Araki, H. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 2007, 445, 328–332. [Google Scholar] [CrossRef]
- Zegerman, P.; Diffley, J.F. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 2007, 445, 281–285. [Google Scholar] [CrossRef]
- Heller, R.C.; Kang, S.; Lam, W.M.; Chen, S.; Chan, C.S.; Bell, S.P. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 2011, 146, 80–91. [Google Scholar] [CrossRef]
- Muramatsu, S.; Hirai, K.; Tak, Y.S.; Kamimura, Y.; Araki, H. CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol (epsilon}, and GINS in budding yeast. Genes Dev. 2010, 24, 602–612. [Google Scholar] [CrossRef]
- Tanaka, T.; Umemori, T.; Endo, S.; Muramatsu, S.; Kanemaki, M.; Kamimura, Y.; Obuse, C.; Araki, H. Sld7, an Sld3-associated protein required for efficient chromosomal DNA replication in budding yeast. EMBO J. 2011, 30, 2019–2030. [Google Scholar] [CrossRef]
- Natsume, T.; Muller, C.A.; Katou, Y.; Retkute, R.; Gierlinski, M.; Araki, H.; Blow, J.J.; Shirahige, K.; Nieduszynski, C.A.; Tanaka, T.U. Kinetochores coordinate pericentromeric cohesion and early DNA replication by Cdc7-Dbf4 kinase recruitment. Mol. Cell 2013, 50, 661–674. [Google Scholar] [CrossRef]
- Dorneiter, I.; Erdile, L.F.; Gilbert, I.U.; von Winkler, D.; Kelly, T.J.; Fanning, E. Interaction of DNA polymerase a-primase with cellular replication protein A and SV40 T antigen. EMBO 1992, 11, 769–776. [Google Scholar]
- Jiang, X.; Klimovich, V.; Arunkumar, A.I.; Hysinger, E.B.; Wang, Y.; Ott, R.D.; Guler, G.D.; Weiner, B.; Chazin, W.J.; Fanning, E. Structural mechanism of RPA loading on DNA during activation of a simple pre-replication complex. EMBO J. 2006, 25, 5516–5526. [Google Scholar] [CrossRef]
- Waga, S.; Stillman, B. The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 1998, 67, 721–751. [Google Scholar] [CrossRef]
- Pursell, Z.F.; Isoz, I.; Lundstrom, E.B.; Johansson, E.; Kunkel, T.A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 2007, 317, 127–130. [Google Scholar] [CrossRef]
- Shikata, K.; Sasa-Masuda, T.; Okuno, Y.; Waga, S.; Sugino, A. The DNA polymerase activity of Pol epsilon holoenzyme is required for rapid and efficient chromosomal DNA replication in Xenopus egg extracts. BMC Biochem. 2006, 7, 21. [Google Scholar] [CrossRef]
- Stillman, B. DNA polymerases at the replication fork in eukaryotes. Mol. Cell 2008, 30, 259–260. [Google Scholar] [CrossRef]
- Maiorano, D.; Cuvier, O.; Danis, E.; Mechali, M. MCM8 is an MCM2-7-related protein that functions as a DNA helicase during replication elongation and not initiation. Cell 2005, 120, 315–328. [Google Scholar] [CrossRef]
- Crevel, G.; Hashimoto, R.; Vass, S.; Sherkow, J.; Yamaguchi, M.; Heck, M.M.; Cotterill, S. Differential requirements for MCM proteins in DNA replication in Drosophila S2 cells. PLoS One 2007, 2, e833. [Google Scholar] [CrossRef]
- Gozuacik, D.; Chami, M.; Lagorce, D.; Faivre, J.; Murakami, Y.; Poch, O.; Biermann, E.; Knippers, R.; Brechot, C.; Paterlini-Brechot, P. Identification and functional characterization of a new member of the human Mcm protein family: hMcm8. Nucleic Acids Res. 2003, 31, 570–579. [Google Scholar] [CrossRef]
- Huberman, J.A.; Riggs, A.D. Autoradiography of chromosomal DNA fibers from Chinese hamster cells. Proc. Natl. Acad. Sci. USA 1966, 55, 599–606. [Google Scholar] [CrossRef]
- Sarkies, P.; Sale, J.E. Cellular epigenetic stability and cancer. Trends Genet. 2012, 28, 118–127. [Google Scholar] [CrossRef]
- Mechali, M. DNA replication origins: From sequence specificity to epigenetics. Nat. Rev. Genet. 2001, 2, 640–645. [Google Scholar] [CrossRef]
- Nishiyama, A.; Frappier, L.; Mechali, M. MCM-BP regulates unloading of the MCM2-7 helicase in late S phase. Genes Dev. 2011, 25, 165–175. [Google Scholar] [CrossRef]
- Rao, P.N.; Johnson, R.T. Mammalian cell fusion: Studies on the regulation of DNA synthesis and mitosis. Nature 1970, 225, 159–164. [Google Scholar] [CrossRef]
- Blow, J.J.; Laskey, R. A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature 1988, 332, 546–548. [Google Scholar] [CrossRef]
- Chong, J.P.; Mahbubani, H.M.; Khoo, C.-Y.; Blow, J.J. Purification of an MCM-containing complex as a component of the DNA replication licensing system. Nature 1995, 375, 418–421. [Google Scholar] [CrossRef]
- Cocker, J.H.; Piatti, S.; Santocanale, C.; Nasmyth, K.; Diffley, J.F.X. An essential role for the Cdc6 protein in forming the pre-replicative complexes of budding yeast. Nature 1996, 379, 180–182. [Google Scholar] [CrossRef]
- Tada, S. Cdt1 and geminin: Role during cell cycle progression and DNA damage in higher eukaryotes. Front. Biosci. 2007, 12, 1629–1641. [Google Scholar] [CrossRef]
- Ballabeni, A.; Zamponi, R.; Moore, J.K.; Helin, K.; Kirschner, M.W. Geminin deploys multiple mechanisms to regulate Cdt1 before cell division thus ensuring the proper execution of DNA replication. Proc. Natl. Acad. Sci. USA 2013, 110, E2848–E2853. [Google Scholar] [CrossRef]
- Maiorano, D.; Moreau, J.; Mechali, M. XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature 2000, 404, 622–625. [Google Scholar] [CrossRef]
- Rialland, M.; Sola, F.; Santocanale, C. Essential role of human CDT1 in DNA replication and chromatin licensing. J. Cell Sci. 2002, 115, 1435–1440. [Google Scholar]
- Nishitani, H.; Taraviras, S.; Lygerou, Z.; Nishimoto, T. The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J. Biol. Chem. 2001, 276, 44905–44911. [Google Scholar] [CrossRef]
- Arias, E.E.; Walter, J.C. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 2006, 8, 84–90. [Google Scholar] [CrossRef]
- Maiorano, D.; Krasinska, L.; Lutzmann, M.; Mechali, M. Recombinant Cdt1 induces rereplication of G2 nuclei in Xenopus egg extracts. Curr. Biol. 2005, 15, 146–153. [Google Scholar] [CrossRef]
- Li, A.; Blow, J.J. Cdt1 downregulation by proteolysis and geminin inhibition prevents DNA re-replication in Xenopus. EMBO J. 2005, 24, 395–404. [Google Scholar] [CrossRef]
- Arias, E.E.; Walter, J.C. Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev. 2005, 19, 114–126. [Google Scholar] [CrossRef]
- Yoshida, K.; Takisawa, H.; Kubota, Y. Intrinsic nuclear import activity of geminin is essential to prevent re-initiation of DNA replication in Xenopus eggs. Genes Cells 2005, 10, 63–73. [Google Scholar] [CrossRef]
- Lutzmann, M.; Maiorano, D.; Mechali, M. A Cdt1-geminin complex licenses chromatin for DNA replication and prevents rereplication during S phase in Xenopus. EMBO J. 2006, 25, 5764–5774. [Google Scholar] [CrossRef]
- Vaziri, C.; Saxena, S.; Jeon, Y.; Lee, C.; Murata, K.; Machida, Y.; Wagle, N.; Hwang, D.S.; Dutta, A. A p53-dependent checkpoint pathway prevents rereplication. Mol. Cell 2003, 11, 997–1008. [Google Scholar] [CrossRef]
- Labib, K.; Diffley, J.F.; Kearsey, S.E. G1-phase and B-type cyclins exclude the DNA-replication factor Mcm4 from the nucleus. Nat. Cell Biol. 1999, 1, 415–422. [Google Scholar]
- Nishitani, H.; Lygerou, Z.; Nishimoto, T.; Nurse, P. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature 2000, 404, 625–628. [Google Scholar] [CrossRef]
- Uhlmann, F.; Bouchoux, C.; Lopez-Aviles, S. A quantitative model for cyclin-dependent kinase control of the cell cycle: Revisited. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3572–3583. [Google Scholar] [CrossRef]
- Starostina, N.G.; Kipreos, E.T. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2011, 22, 33–41. [Google Scholar] [CrossRef]
- Santamaria, D.; Barriere, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Caceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef]
- Fisher, D.; Nurse, P. A single fission yeast mitotic cyclin B-p34cdc2 kinase promotes both S-phase and mitosis in the absence of G1-cyclins. EMBO J. 1996, 15, 850–860. [Google Scholar]
- Sansam, C.L.; Cruz, N.M.; Danielian, P.S.; Amsterdam, A.; Lau, M.L.; Hopkins, N.; Lees, J.A. A vertebrate gene, ticrr, is an essential checkpoint and replication regulator. Genes Dev. 2010, 24, 183–194. [Google Scholar] [CrossRef]
- Kumagai, A.; Shevchenko, A.; Dunphy, W.G. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef]
- Balestrini, A.; Cosentino, C.; Errico, A.; Garner, E.; Costanzo, V. GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nat. Cell Biol. 2010, 12, 484–491. [Google Scholar] [CrossRef]
- Wang, Z.; Kim, E.; Leffak, M.; Xu, Y.J. Treslin, DUE-B, and GEMC1 cannot complement Sld3 mutants in fission yeast. FEMS Yeast Res. 2012, 12, 486–490. [Google Scholar] [CrossRef]
- Stern, B.; Nurse, P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends Genet. 1996, 12, 345–350. [Google Scholar] [CrossRef]
- Koundrioukoff, S.; Jonsson, Z.O.; Hasan, S.; de Jong, R.N.; van der Vliet, P.C.; Hottiger, M.O.; Hubscher, U. A direct interaction between proliferating cell nuclear antigen (PCNA) and Cdk2 targets PCNA-interacting proteins for phosphorylation. J. Biol. Chem. 2000, 275, 22882–22887. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Lambert, S.; Carr, A.M. Checkpoint responses to replication fork barriers. Biochimie 2005, 87, 591–602. [Google Scholar] [CrossRef]
- Rupp, W.D.; Howard-Flanders, P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J. Mol. Biol. 1968, 31, 291–304. [Google Scholar] [CrossRef]
- Pages, V.; Fuchs, R.P. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science 2003, 300, 1300–1303. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar]
- De Klein, A.; Muijtjens, M.; van Os, R.; Verhoeven, Y.; Smit, B.; Carr, A.M.; Lehmann, A.R.; Hoeijmakers, J.H. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000, 10, 479–482. [Google Scholar] [CrossRef]
- Daniel, J.A.; Pellegrini, M.; Lee, B.S.; Guo, Z.; Filsuf, D.; Belkina, N.V.; You, Z.; Paull, T.T.; Sleckman, B.P.; Feigenbaum, L.; et al. Loss of ATM kinase activity leads to embryonic lethality in mice. J. Cell Biol. 2012, 198, 295–304. [Google Scholar] [CrossRef]
- Yamamoto, K.; Wang, Y.; Jiang, W.; Liu, X.; Dubois, R.L.; Lin, C.S.; Ludwig, T.; Bakkenist, C.J.; Zha, S. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J. Cell Biol. 2012, 198, 305–313. [Google Scholar] [CrossRef]
- Zegerman, P.; Diffley, J.F. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef]
- Mailand, N.; Falck, J.; Lukas, C.; Syljuasen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid destruction of human Cdc25A in response to DNA damage. Science 2000, 288, 1425–1429. [Google Scholar] [CrossRef]
- Costanzo, V.; Robertson, K.; Ying, C.Y.; Kim, E.; Avvedimento, E.; Gottesman, M.; Grieco, D.; Gautier, J. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol. Cell 2000, 6, 649–659. [Google Scholar] [CrossRef]
- Tsuji, T.; Lau, E.; Chiang, G.G.; Jiang, W. The role of Dbf4/Drf1-dependent kinase Cdc7 in DNA-damage checkpoint control. Mol. Cell 2008, 32, 862–869. [Google Scholar] [CrossRef]
- Lee, A.Y.; Chiba, T.; Truong, L.N.; Cheng, A.N.; Do, J.; Cho, M.J.; Chen, L.; Wu, X. Dbf4 is direct downstream target of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) protein to regulate intra-S-phase checkpoint. J. Biol. Chem. 2012, 287, 2531–2543. [Google Scholar]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef]
- Lonn, U.; Lonn, S. Extensive regions of single-stranded DNA in aphidicolin-treated melanoma cells. Biochemistry 1988, 27, 566–570. [Google Scholar] [CrossRef]
- Gobbini, E.; Cesena, D.; Galbiati, A.; Lockhart, A.; Longhese, M.P. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair 2013, 12, 791–799. [Google Scholar] [CrossRef]
- D’Amours, D.; Jackson, S.P. The Mre11 complex: At the crossroads of dna repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 317–327. [Google Scholar] [CrossRef]
- Willis, J.; Patel, Y.; Lentz, B.L.; Yan, S. APE2 is required for ATR-Chk1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 10592–10597. [Google Scholar] [CrossRef]
- Gamper, A.M.; Choi, S.; Matsumoto, Y.; Banerjee, D.; Tomkinson, A.E.; Bakkenist, C.J. ATM protein physically and functionally interacts with proliferating cell nuclear antigen to regulate DNA synthesis. J. Biol. Chem. 2012, 287, 12445–12454. [Google Scholar] [CrossRef]
- Costanzo, V.; Shechter, D.; Lupardus, P.J.; Cimprich, K.A.; Gottesman, M.; Gautier, J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol. Cell 2003, 11, 203–213. [Google Scholar] [CrossRef]
- Zou, L.; Liu, D.; Elledge, S.J. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 13827–13832. [Google Scholar] [CrossRef]
- Ball, H.L.; Myers, J.S.; Cortez, D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol. Biol. Cell 2005, 16, 2372–2381. [Google Scholar] [CrossRef]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef]
- Dart, D.A.; Adams, K.E.; Akerman, I.; Lakin, N.D. Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J. Biol. Chem. 2004, 279, 16433–16440. [Google Scholar]
- Recolin, B.; van der Laan, S.; Maiorano, D. Role of replication protein A as sensor in activation of the S-phase checkpoint in Xenopus egg extracts. Nucleic Acids Res. 2012, 40, 3431–3442. [Google Scholar] [CrossRef]
- Dodson, G.E.; Shi, Y.; Tibbetts, R.S. DNA replication defects, spontaneous DNA damage, and ATM-dependent checkpoint activation in replication protein A-deficient cells. J. Biol. Chem. 2004, 279, 34010–34014. [Google Scholar] [CrossRef]
- Lucca, C.; Vanoli, F.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Haber, J.; Foiani, M. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 2004, 23, 1206–1213. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Ashton, N.W.; Bolderson, E.; Cubeddu, L.; O’Byrne, K.J.; Richard, D.J. Human single-stranded DNA binding proteins are essential for maintaining genomic stability. BMC Mol. Biol. 2013, 14, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Putnam, C.D.; Kane, M.F.; Zhang, W.; Edelmann, L.; Russell, R.; Carrion, D.V.; Chin, L.; Kucherlapati, R.; Kolodner, R.D.; et al. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat. Genet. 2005, 37, 750–755. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef]
- Bermudez, V.P.; Lindsey-Boltz, L.A.; Cesare, A.J.; Maniwa, Y.; Griffith, J.D.; Hurwitz, J.; Sancar, A. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 1633–1638. [Google Scholar] [CrossRef]
- Ellison, V.; Stillman, B. Biochemical characterization of DNA damage checkpoint complexes: Clamp loader and clamp complexes with specificity for 5' recessed DNA. PLoS Biol. 2003, 1, E33. [Google Scholar]
- Zou, L.; Cortez, D.; Elledge, S.J. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002, 16, 198–208. [Google Scholar] [CrossRef]
- MacDougall, C.A.; Byun, T.S.; Van, C.; Yee, M.C.; Cimprich, K.A. The structural determinants of checkpoint activation. Genes Dev. 2007, 21, 898–903. [Google Scholar] [CrossRef]
- Majka, J.; Binz, S.K.; Wold, M.S.; Burgers, P.M. Replication protein A directs loading of the DNA damage checkpoint clamp to 5'-DNA junctions. J. Biol. Chem. 2006, 281, 27855–27861. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Takisawa, H. Xenopus Cut5 is essential for a CDK-dependent process in the initiation of DNA replication. EMBO J. 2003, 22, 2526–2535. [Google Scholar] [CrossRef]
- Parrilla-Castellar, E.R.; Karnitz, L.M. Cut5 is required for the binding of Atr and DNA polymerase alpha to genotoxin-damaged chromatin. J. Biol. Chem. 2003, 278, 45507–45511. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Tsujimura, T.; Sugino, A.; Takisawa, H. The phosphorylated C-terminal domain of Xenopus Cut5 directly mediates ATR-dependent activation of Chk1. Genes Cells 2006, 11, 993–1007. [Google Scholar] [CrossRef]
- Yan, S.; Lindsay, H.D.; Michael, W.M. Direct requirement for Xmus101 in ATR-mediated phosphorylation of Claspin bound Chk1 during checkpoint signaling. J. Cell Biol. 2006, 173, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Handa, T.; Kanke, M.; Takahashi, T.S.; Nakagawa, T.; Masukata, H. DNA polymerization-independent functions of DNA polymerase epsilon in assembly and progression of the replisome in fission yeast. Mol. Biol. Cell 2012, 23, 3240–3253. [Google Scholar] [CrossRef]
- Delacroix, S.; Wagner, J.M.; Kobayashi, M.; Yamamoto, K.; Karnitz, L.M. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007, 21, 1472–1477. [Google Scholar]
- Lee, J.; Kumagai, A.; Dunphy, W.G. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 2007, 282, 28036–28044. [Google Scholar] [CrossRef]
- Furuya, K.; Poitelea, M.; Guo, L.; Caspari, T.; Carr, A.M. Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 2004, 18, 1154–1164. [Google Scholar] [CrossRef]
- Mordes, D.A.; Glick, G.G.; Zhao, R.; Cortez, D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008, 22, 1478–1489. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Yan, S.; Michael, W.M. TopBP1 and DNA polymerase alpha-mediated recruitment of the 9-1-1 complex to stalled replication forks: Implications for a replication restart-based mechanism for ATR checkpoint activation. Cell Cycle 2009, 8, 2877–2884. [Google Scholar] [CrossRef]
- Gong, Z.; Kim, J.E.; Leung, C.C.; Glover, J.N.; Chen, J. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol. Cell 2010, 37, 438–446. [Google Scholar] [CrossRef]
- Wang, J.; Gong, Z.; Chen, J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J. Cell Biol. 2011, 193, 267–273. [Google Scholar] [CrossRef]
- Duursma, A.M.; Driscoll, R.; Elias, J.E.; Cimprich, K.A. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol. Cell 2013, 50, 116–122. [Google Scholar] [CrossRef]
- Lee, J.; Dunphy, W.G. The Mre11-Rad50-Nbs1 (MRN) complex has a specific role in the activation of Chk1 in response to stalled replication forks. Mol. Biol. Cell 2013, 24, 1343–1353. [Google Scholar] [CrossRef]
- Michael, W.M.; Ott, R.; Fanning, E.; Newport, J. Activation of the DNA replication checkpoint through RNA synthesis by primase. Science 2000, 289, 2133–2137. [Google Scholar] [CrossRef]
- Lupardus, P.J.; Byun, T.; Yee, M.C.; Hekmat-Nejad, M.; Cimprich, K.A. A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev. 2002, 16, 2327–2332. [Google Scholar] [CrossRef]
- Van, C.; Yan, S.; Michael, W.M.; Waga, S.; Cimprich, K.A. Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J. Cell Biol. 2010, 189, 233–246. [Google Scholar] [CrossRef]
- Liu, S.; Shiotani, B.; Lahiri, M.; Marechal, A.; Tse, A.; Leung, C.C.; Glover, J.N.; Yang, X.H.; Zou, L. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell 2011, 43, 192–202. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Jazayeri, A.; Balestrini, A.; Garner, E.; Haber, J.E.; Costanzo, V. Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. EMBO J. 2008, 27, 1953–1962. [Google Scholar] [CrossRef]
- Helleday, T. PrimPol breaks replication barriers. Nat. Struct. Mol. Biol. 2013, 20, 1348–1350. [Google Scholar] [CrossRef]
- Bianchi, J.; Rudd, S.G.; Jozwiakowski, S.K.; Bailey, L.J.; Soura, V.; Taylor, E.; Stevanovic, I.; Green, A.J.; Stracker, T.H.; Lindsay, H.D.; et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell 2013, 52, 566–573. [Google Scholar] [CrossRef]
- Mouron, S.; Rodriguez-Acebes, S.; Martinez-Jimenez, M.I.; Garcia-Gomez, S.; Chocron, S.; Blanco, L.; Mendez, J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013, 14, 1104–1112. [Google Scholar] [CrossRef]
- Garcia-Gomez, S.; Reyes, A.; Martinez-Jimenez, M.I.; Chocron, E.S.; Mouron, S.; Terrados, G.; Powell, C.; Salido, E.; Mendez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef]
- Gambus, A.; van Deursen, F.; Polychronopoulos, D.; Foltman, M.; Jones, R.C.; Edmondson, R.D.; Calzada, A.; Labib, K. A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J. 2009, 28, 2992–3004. [Google Scholar] [CrossRef]
- Katou, Y.; Kanoh, Y.; Bando, M.; Noguchi, H.; Tanaka, H.; Ashikari, T.; Sugimoto, K.; Shirahige, K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 2003, 424, 1078–1083. [Google Scholar] [CrossRef]
- Hekmat-Nejad, M.; You, Z.; Yee, M.C.; Newport, J.W.; Cimprich, K.A. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr. Biol. 2000, 10, 1565–1573. [Google Scholar] [CrossRef]
- Bonilla, C.Y.; Melo, J.A.; Toczyski, D.P. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol. Cell 2008, 30, 267–276. [Google Scholar] [CrossRef]
- Melo, J.A.; Cohen, J.; Toczyski, D.P. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev. 2001, 15, 2809–2821. [Google Scholar]
- Soutoglou, E.; Misteli, T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science 2008, 320, 1507–1510. [Google Scholar] [CrossRef]
- Berens, T.J.; Toczyski, D.P. Colocalization of Mec1 and Mrc1 is sufficient for Rad53 phosphorylation in vivo. Mol. Biol. Cell 2012, 23, 1058–1067. [Google Scholar] [CrossRef]
- Betous, R.; Pillaire, M.J.; Pierini, L.; van der Laan, S.; Recolin, B.; Ohl-Seguy, E.; Guo, C.; Niimi, N.; Gruz, P.; Nohmi, T.; et al. DNA polymerase kappa-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J. 2013, 32, 2172–2185. [Google Scholar] [CrossRef]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar]
- Ishimi, Y.; Komamura-Kohno, Y.; Kwon, H.J.; Yamada, K.; Nakanishi, M. Identification of MCM4 as a target of the DNA replication block checkpoint system. J. Biol. Chem. 2003, 278, 24644–24650. [Google Scholar]
- Shechter, D.; Gautier, J. MCM proteins and checkpoint kinases get together at the fork. Proc. Natl. Acad. Sci. USA 2004, 101, 10845–10846. [Google Scholar] [CrossRef]
- Robison, J.G.; Elliott, J.; Dixon, K.; Oakley, G.G. Replication protein A and the Mre11.Rad50.Nbs1 complex co-localize and interact at sites of stalled replication forks. J. Biol. Chem. 2004, 279, 34802–34810. [Google Scholar] [CrossRef]
- Carty, M.P.; Zernik-Kobak, M.; McGrath, S.; Dixon, K. UV light-induced DNA synthesis arrest in HeLa cells is associated with changes in phosphorylation of human single-stranded DNA-binding protein. EMBO J. 1994, 13, 2114–2123. [Google Scholar]
- Shao, R.G.; Cao, C.X.; Zhang, H.; Kohn, K.W.; Wold, M.S.; Pommier, Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999, 18, 1397–1406. [Google Scholar] [CrossRef]
- Recolin, B.; Maiorano, D. Implication of RPA32 phosphorylation in S-phase checkpoint signalling at replication forks stalled with aphidicolin in Xenopus egg extracts. Biochem. Biophys. Res. Commun. 2012, 427, 785–789. [Google Scholar] [CrossRef]
- Lindsey-Boltz, L.A.; Reardon, J.T.; Wold, M.S.; Sancar, A. In vitro analysis of the role of RPA and RPA phosphorylation in ATR-mediated checkpoint signaling. J. Biol. Chem. 2012, 287, 36123–36131. [Google Scholar] [CrossRef]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef]
- Kumagai, A.; Guo, Z.; Emami, K.H.; Wang, S.X.; Dunphy, W.G. The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J. Cell Biol. 1998, 142, 1559–1569. [Google Scholar] [CrossRef]
- Lee, J.; Kumagai, A.; Dunphy, W.G. Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol. Cell 2003, 11, 329–340. [Google Scholar] [CrossRef]
- Naylor, M.L.; Li, J.M.; Osborn, A.J.; Elledge, S.J. Mrc1 phosphorylation in response to DNA replication stress is required for Mec1 accumulation at the stalled fork. Proc. Natl. Acad. Sci. USA 2009, 106, 12765–12770. [Google Scholar] [CrossRef]
- Lin, S.Y.; Li, K.; Stewart, G.S.; Elledge, S.J. Human Claspin works with BRCA1 to both positively and negatively regulate cell proliferation. Proc. Natl. Acad. Sci. USA 2004, 101, 6484–6489. [Google Scholar] [CrossRef]
- Unsal-Kacmaz, K.; Chastain, P.D.; Qu, P.P.; Minoo, P.; Cordeiro-Stone, M.; Sancar, A.; Kaufmann, W.K. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 2007, 27, 3131–3142. [Google Scholar] [CrossRef]
- Numata, Y.; Ishihara, S.; Hasegawa, N.; Nozaki, N.; Ishimi, Y. Interaction of human MCM2-7 proteins with TIM, TIPIN and Rb. J. Biochem. 2010, 147, 917–927. [Google Scholar] [CrossRef]
- Kemp, M.G.; Akan, Z.; Yilmaz, S.; Grillo, M.; Smith-Roe, S.L.; Kang, T.H.; Cordeiro-Stone, M.; Kaufmann, W.K.; Abraham, R.T.; Sancar, A.; et al. Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J. Biol. Chem. 2010, 285, 16562–16571. [Google Scholar] [CrossRef]
- Lee, J.; Gold, D.A.; Shevchenko, A.; Dunphy, W.G. Roles of replication fork-interacting and Chk1-activating domains from claspin in a DNA replication checkpoint response. Mol. Biol. Cell 2005, 16, 5269–5282. [Google Scholar] [CrossRef]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar]
- Lopez-Girona, A.; Tanaka, K.; Chen, X.B.; Baber, B.A.; McGowan, C.H.; Russell, P. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc. Natl. Acad. Sci. USA 2001, 98, 11289–11294. [Google Scholar] [CrossRef]
- Santocanale, C.; Diffley, J.F. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 1998, 395, 615–618. [Google Scholar] [CrossRef]
- Shechter, D.; Costanzo, V.; Gautier, J. ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell Biol. 2004, 6, 648–655. [Google Scholar] [CrossRef]
- Woodward, A.M.; Gohler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef]
- De Piccoli, G.; Katou, Y.; Itoh, T.; Nakato, R.; Shirahige, K.; Labib, K. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol. Cell 2012, 45, 696–704. [Google Scholar] [CrossRef]
- Blasius, M.; Forment, J.V.; Thakkar, N.; Wagner, S.A.; Choudhary, C.; Jackson, S.P. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 2011, 12, R78. [Google Scholar] [CrossRef]
- Jinno, S.; Suto, K.; Nagata, A.; Igarashi, M.; Kanaoka, Y.; Nojima, H.; Okayama, H. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 1994, 13, 1549–1556. [Google Scholar]
- Molinari, M.; Mercurio, C.; Dominguez, J.; Goubin, F.; Draetta, G.F. Human Cdc25 A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Rep. 2000, 1, 71–79. [Google Scholar] [CrossRef]
- Donzelli, M.; Squatrito, M.; Ganoth, D.; Hershko, A.; Pagano, M.; Draetta, G.F. Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 2002, 21, 4875–4884. [Google Scholar] [CrossRef]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef]
- Van der Laan, S.; Tsanov, N.; Crozet, C.; Maiorano, D. High Dub3 expression in mouse ESCs couples the G1/S checkpoint to pluripotency. Mol. Cell 2013, 52, 366–379. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2011, 29, 5095–5102. [Google Scholar]
- Neelsen, K.J.; Zanini, I.M.; Herrador, R.; Lopes, M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013, 200, 699–708. [Google Scholar] [CrossRef] [Green Version]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Recolin, B.; Van der Laan, S.; Tsanov, N.; Maiorano, D. Molecular Mechanisms of DNA Replication Checkpoint Activation. Genes 2014, 5, 147-175. https://doi.org/10.3390/genes5010147
Recolin B, Van der Laan S, Tsanov N, Maiorano D. Molecular Mechanisms of DNA Replication Checkpoint Activation. Genes. 2014; 5(1):147-175. https://doi.org/10.3390/genes5010147
Chicago/Turabian StyleRecolin, Bénédicte, Siem Van der Laan, Nikolay Tsanov, and Domenico Maiorano. 2014. "Molecular Mechanisms of DNA Replication Checkpoint Activation" Genes 5, no. 1: 147-175. https://doi.org/10.3390/genes5010147
APA StyleRecolin, B., Van der Laan, S., Tsanov, N., & Maiorano, D. (2014). Molecular Mechanisms of DNA Replication Checkpoint Activation. Genes, 5(1), 147-175. https://doi.org/10.3390/genes5010147