The Genomic Signature of Breast Cancer Prevention


Abstract
:1. Introduction
2. Phenotypic Changes Induced by Pregnancy in the Human Breast

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Log Ratio | P value | Gene Name |
|---|---|---|---|
| Apoptosis (GO:0006915; GO:0006917; GO:0008624; GO:0042981) | |||
| CASP4 | 0.37 | 0.0003 | caspase 4, apoptosis-related cysteine peptidase |
| RUNX3 | 0.36 | 0.0000 | runt-related transcription factor 3 |
| LUC7L3 | 0.34 | 0.0002 | LUC7-like 3 (S. cerevisiae) |
| ELMO3 | 0.30 | 0.0003 | engulfment and cell motility 3 |
| DNA repair (GO:0006281; GO:0006284) | |||
| SFPQ | 0.46 | 0.0002 | splicing factor proline/glutamine-rich |
| MBD4 | 0.36 | 0.0003 | methyl-CpG binding domain protein 4 |
| RBBP8 | 0.32 | 0.0000 | retinoblastoma binding protein 8 |
| Cell adhesion (GO:0007155; GO:0030155) | |||
| NRXN1 | 0.60 | 0.0001 | neurexin 1 |
| DSC3 | 0.51 | 0.0000 | desmocollin 3 |
| COL27A1 | 0.44 | 0.0002 | collagen, type XXVII, alpha 1 |
| PNN | 0.37 | 0.0001 | pinin, desmosome associated protein |
| COL4A6 | 0.36 | 0.0008 | collagen, type IV, alpha 6 |
| LAMC2 | 0.34 | 0.0008 | laminin, gamma 2 |
| COL7A1 | 0.33 | 0.0002 | collagen, type VII, alpha 1 |
| COL16A1 | 0.31 | 0.0000 | collagen, type XVI, alpha 1 |
| LAMA3 | 0.30 | 0.0008 | laminin, alpha 3 |
| Cell cycle (GO:0000075; GO:0007049; GO:0045786) | |||
| SYCP2 | 0.45 | 0.0000 | synaptonemal complex protein 2 |
| PNN | 0.37 | 0.0001 | pinin, desmosome associated protein |
| RUNX3 | 0.36 | 0.0000 | runt-related transcription factor 3 |
| RBBP8 | 0.32 | 0.0000 | retinoblastoma binding protein 8 |
| Cell differentiation (GO:0001709; GO:0030154; GO:0030216) | |||
| MGP | 0.53 | 0.0003 | matrix Gla protein |
| KRT5 | 0.41 | 0.0002 | keratin 5 |
| GATA3 | 0.35 | 0.0009 | GATA binding protein 3 |
| LAMA3 | 0.30 | 0.0008 | laminin, alpha 3 |
| Cell proliferation (GO:0008283; GO:0008284; GO:0008285; GO:0042127; GO:0050679; GO:0050680) | |||
| PTN | 0.67 | 0.0002 | Pleiotrophin |
| KRT5 | 0.41 | 0.0002 | keratin 5 |
| RUNX3 | 0.36 | 0.0000 | runt-related transcription factor 3 |
| IL28RA | 0.34 | 0.0003 | interleukin 28 receptor, alpha (interferon, lambda receptor) |
| CDCA7 | 0.31 | 0.0005 | cell division cycle associated 7 |
| Cell motility (GO:0006928; GO:0030334) | |||
| DNALI1 | 0.37 | 0.0001 | dynein, axonemal, light intermediate chain 1 |
| LAMA3 | 0.30 | 0.0008 | laminin, alpha 3 |
| G-protein coupled receptor pathway (GO:0007186) | |||
| OXTR | 0.54 | 0.0006 | oxytocin receptor |
| RNA metabolic process (GO:0000398; GO:0001510; GO:0006376; GO:0006396; GO:0006397; GO:0006401; GO:0008380) | |||
| METTL3 | 0.69 | 0.0000 | methyltransferase like 3 |
| HNRPDL | 0.65 | 0.0001 | heterogeneous nuclear ribonucleoprotein D-like |
| HNRNPD | 0.59 | 0.0003 | heterogeneous nuclear ribonucleoprotein D (AU-rich element RNA binding protein 1, 37 kDa) |
| HNRNPA2B1 | 0.56 | 0.0003 | heterogeneous nuclear ribonucleoprotein A2/B1 |
| SFPQ | 0.47 | 0.0006 | splicing factor proline/glutamine-rich |
| RBM25 | 0.38 | 0.0009 | RNA binding motif protein 25 |
| RBMX | 0.38 | 0.0000 | RNA binding motif protein, X-linked |
| LUC7L3 | 0.34 | 0.0002 | LUC7-like 3 (S. cerevisiae) |
| SFRS1 | 0.30 | 0.0001 | splicing factor, arginine/serine-rich 1 |
| RNA transport (GO:0050658) | |||
| HNRNPA2B1 | 0.56 | 0.0003 | heterogeneous nuclear ribonucleoprotein A2/B1 |
| Transcription (GO:0006350; GO:0006355; GO:0006357; GO:0006366; GO:0016481; GO:0045449; GO:0045893; GO:0045941) | |||
| HNRPDL | 0.65 | 0.0001 | heterogeneous nuclear ribonucleoprotein D-like |
| HNRNPD | 0.59 | 0.0003 | heterogeneous nuclear ribonucleoprotein D (AU-rich element RNA binding protein 1, 37 kDa) |
| CBX3 | 0.53 | 0.0003 | chromobox homolog 3 (HP1 gamma homolog, Drosophila) |
| NFKBIZ | 0.48 | 0.0001 | nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, zeta |
| FUBP1 | 0.47 | 0.0002 | far upstream element (FUSE) binding protein 1 |
| SFPQ | 0.47 | 0.0006 | splicing factor proline/glutamine-rich |
| EZH2 | 0.44 | 0.0000 | enhancer of zeste homolog 2 (Drosophila) |
| ZNF207 | 0.41 | 0.0007 | zinc finger protein 207 |
| ZNF711 | 0.41 | 0.0003 | zinc finger protein 711 |
| GATA3 | 0.38 | 0.0009 | GATA binding protein 3 |
| PNN | 0.37 | 0.0003 | pinin, desmosome associated protein |
| ZNF107 | 0.37 | 0.0001 | zinc finger protein 107 |
| RUNX3 | 0.36 | 0.0000 | runt-related transcription factor 3 |
| CCNL1 | 0.35 | 0.0009 | cyclin L1 |
| ZNF692 | 0.34 | 0.0000 | zinc finger protein 692 |
| CHD2 | 0.33 | 0.0001 | chromodomain helicase DNA binding protein 2 |
| RBBP8 | 0.32 | 0.0000 | retinoblastoma binding protein 8 |
| ZNF789 | 0.32 | 0.0005 | zinc finger protein 789 |
| CDCA7 | 0.31 | 0.0005 | cell division cycle associated 7 |
| Chromatin organization (GO:0006333; GO:0006338) | |||
| CBX3 | 0.53 | 0.0003 | chromobox homolog 3 (HP1 gamma homolog, Drosophila) |
| CHD2 | 0.33 | 0.0001 | chromodomain helicase DNA binding protein 2 |
| Cell division (GO:0051301) | |||
| SYCP2 | 0.45 | 0.0000 | synaptonemal complex protein 2 |
| DNA metabolic process (GO:0006139; GO:0006260; GO:0006310; GO:0015074) | |||
| METTL3 | 0.69 | 0.0000 | methyltransferase like 3 |
| SFPQ | 0.46 | 0.0002 | splicing factor proline/glutamine-rich |
| GOLGA2B | 0.32 | 0.0001 | golgin A2 family, member B |
| Lactation (GO:0007595) | |||
| OXTR | 0.54 | 0.0006 | oxytocin receptor |
3. Transcriptomic Differences Induced by Pregnancy
4. Shifting of the Cell Population in the Human Breast
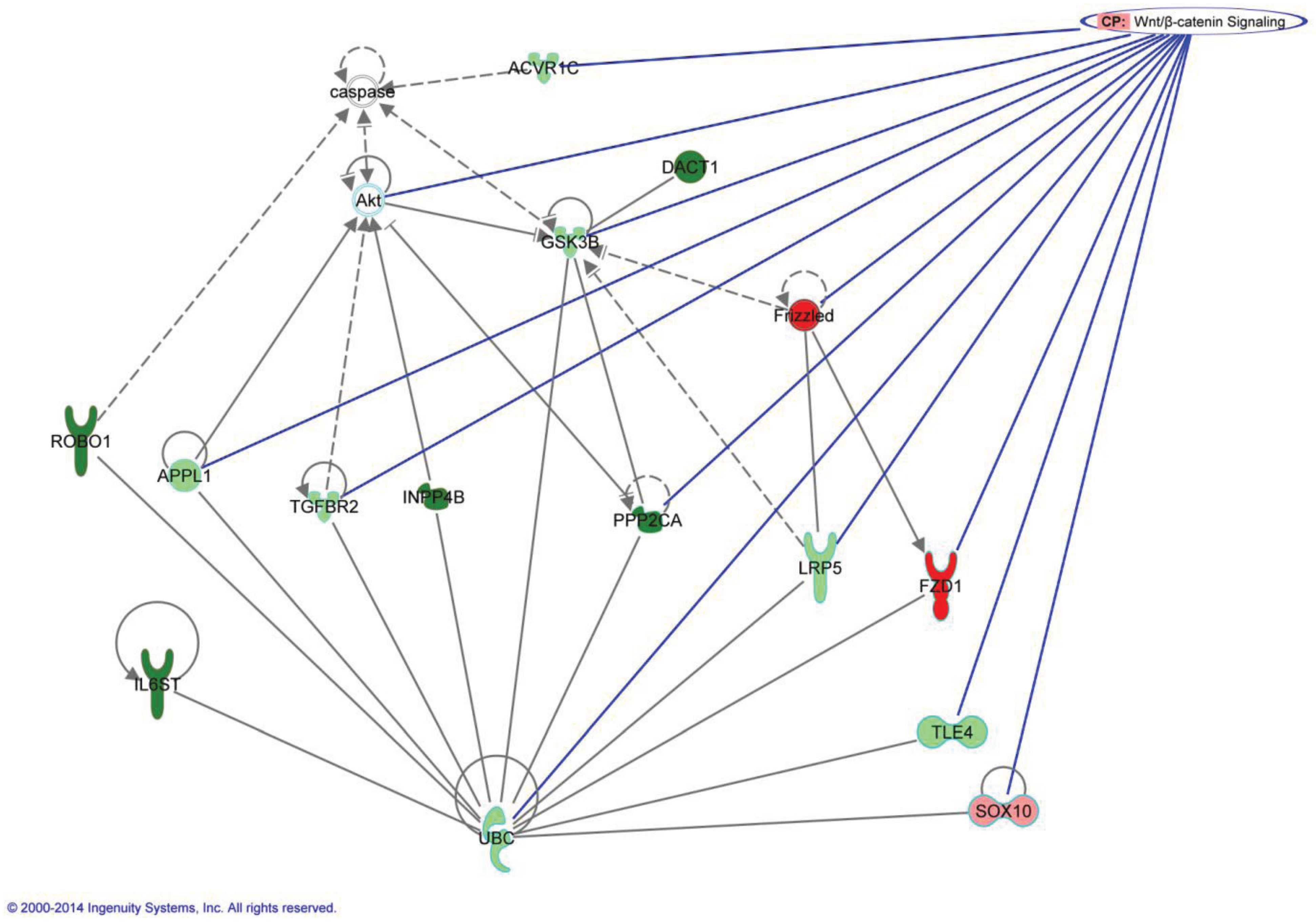
5. Methylation Changes in the DNA of Parous Women are Part of Chromatin Remodeling and the Genomic Signature of Pregnancy


| Parous Hypermethylated Genes | ||
|---|---|---|
| NEGR1 | chr1 | 71702567-71703327 |
| NUF2 | chr1 | 161576182-161576653 |
| SYT14 | chr1 | 208309959-208310406 |
| POU4F1 | chr13 | 78072725-78073146 |
| FLRT2 | chr14 | 85155301-85155789 |
| ASAP2 | chr2 | 9266977-9267464 |
| DNAJC13 | Chr3 | 133712540-133712930 |
| IFITM4P | Chr6 | 29826792-29827266 |
| ZNF292 | Chr6 | 88022117-88022631 |
| SDK1 | Chr7 | 4121961-4122279 |
| ELAVL4 | Chr1 | 50387715-50388146 |
| DACT1 | Chr14 | 58182547-58182717 |
| SPATA5L1 | Chr15 | 43494615-43495210 |
| DYNC1I2 | Chr2 | 172279940-172280462 |
| NLGN1 | Chr3 | 175147546-175148159 |
| MAN1A1 | Chr6 | 119623891-119624320 |
| AK5 | Chr1 | 77616541-77616886 |
| DPYD | Chr1 | 98153997-98154252 |
| PROX1 | Chr1 | 212267523-212267905 |
| PDE3A | Chr12 | 20432463-20432808 |
| NOVA1 | Chr14 | 26015695-26016215 |
| SKAP1 | Chr17 | 43591761-43592022 |
| ANKRD12 | Chr18 | 9168269-9168654 |
| B4GALT5 | Chr20 | 47704095-47704520 |
| CNTN4 | Chr3 | 2572819-2573349 |
| ROBO1 | Chr3 | 79026030-79023709 |
| GSK3B | Chr3 | 121258375-121258501 |
| INPP4B | Chr4 | 143292977-143293319 |
| FNIP2 | Chr4 | 159911129-159911596 |
| IL6ST | Chr5 | 55271135-55271466 |
| TICAM2 | Chr5 | 114955685-114955992 |
| PPP2CA | Chr5 | 133567556-133567871 |
| C6orf138 | Chr6 | 48025616-48025836 |
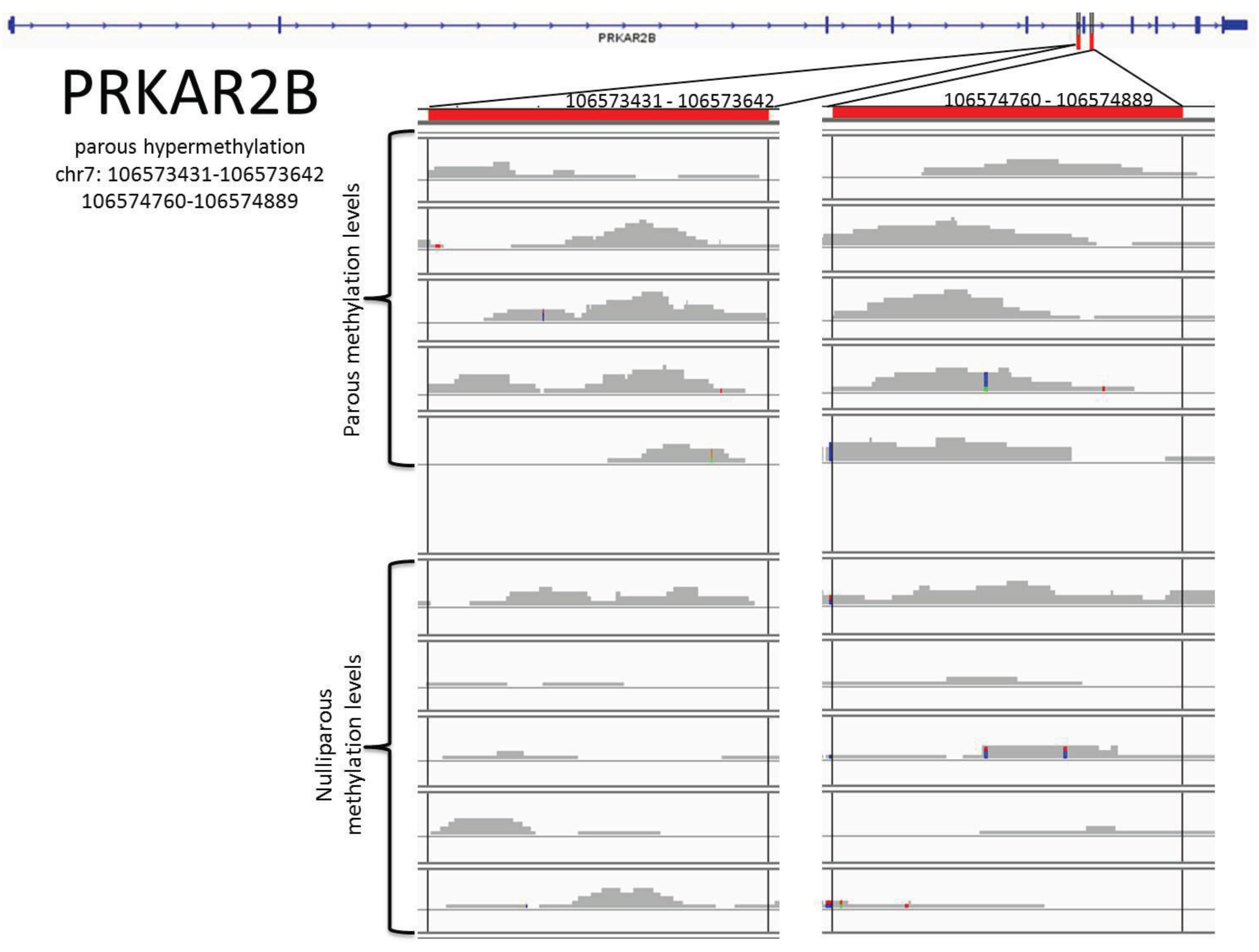
| PRKAR2B | Chr7 | 106573431-106573642 |
| TTLL7 | Chr1 | 84185339-84185660 |
| MAN1A2 | Chr1 | 117816180-117816444 |
| CDC42BPA | Chr1 | 225520202-225520399 |
| OSBP | Chr11 | 59121100-59121437 |
| STIM2 | Chr4 | 26572404-26572775 |
| NR3C2 | Chr4 | 149367631-149368052 |
| REV3L | Chr6 | 111804054-111804285 |
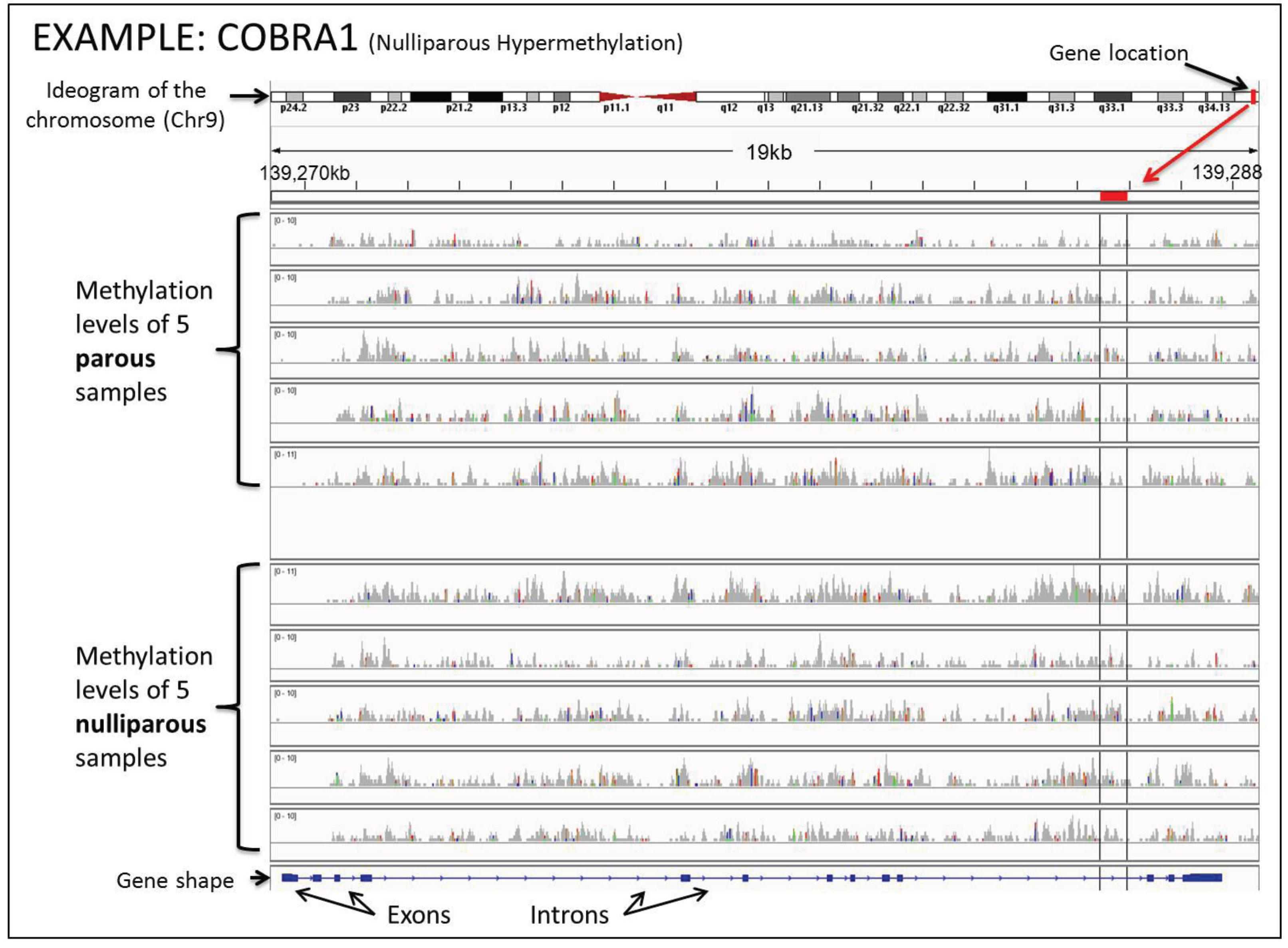
| Nulliparous Hypermethylated Genes | ||
|---|---|---|
| NHSL2 | chrX | 71270541-71271527 |
| C16orf38 (PTX4) | Chr16 | 1476600-1476773 |
| LRRC37A3 | Chr17 | 60311872-60311982 |
| C20orf200 (C20orf166-AS1) | Chr20 | 60557111-60557421 |
| TPPP | Chr5 | 742334-742618 |
| NELF | Chr9 | 139471353-139471653 (HYPO) |
| 139471653-139471895 | ||
| SAMD10 | Chr20 | 62077471-62077661 |
| CELSR1 | Chr22 | 45272965-45273071 |
| FZD1 | Chr7 | 90733372-90733621 |
| TNFRSF18 | Chr1 | 1130349-1130634 |
| SRMS | Chr20 | 61646714-61647041 |
| COBRA1 | Chr9 | 139285424-139285977 |


6. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clarke, C.A.; Purdie, D.M.; Glaser, S.L. Population attributable risk of breast cancer in white women associated with immediately modifiable risk factors. BMC Cancer 2006, 6, 170–181. [Google Scholar] [CrossRef]
- MacMahon, B.; Cole, P.; Lin, T.M.; Lowe, C.R.; Mirra, A.P.; Ravnihar, B.; Salber, E.J.; Valaoras, V.G.; Yuasa, S. Age at first birth and breast cancer risk. Bull. World Health Organ. 1970, 43, 209–221. [Google Scholar]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer statistics, 2007. CA Cancer J. Clin. 2007, 57, 43–66. [Google Scholar] [CrossRef]
- Russo, J.; Balogh, G.A.; Russo, I.H. Full-term pregnancy induces a specific genomic signature in the human breast. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 51–66. [Google Scholar] [CrossRef]
- Russo, J.; Russo, I.H. Influence of differentiation and cell kinetics on the susceptibility of the rat mammary gland to carcinogenesis. Cancer Res. 1980, 40, 2677–2687. [Google Scholar]
- Tay, L.K.; Russo, J. Formation and removal of 7,12-dimethylbenz[a]anthracene–nucleic acid adducts in rat mammary epithelial cells with different susceptibility to carcinogenesis. Carcinogenesis 1981, 2, 1327–1333. [Google Scholar] [CrossRef]
- Russo, I.H.; Koszalka, M.; Russo, J. Comparative study of the influence of pregnancy and hormonal treatment on mammary carcinogenesis. Br. J. Cancer 1991, 64, 481–484. [Google Scholar] [CrossRef]
- Sinha, D.K.; Pazik, J.E.; Dao, T.L. Prevention of mammary carcinogenesis in rats by pregnancy: Effect of full-term and interrupted pregnancy. Br. J. Cancer 1988, 57, 390–394. [Google Scholar] [CrossRef]
- Srivastava, P.; Russo, J.; Mgbonyebi, O.P.; Russo, I.H. Growth inhibition and activation of apoptotic gene expression by human chorionic gonadotropin in human breast epithelial cells. Anticancer Res. 1998, 18, 4003–4010. [Google Scholar]
- Thomas, D.B.; Rosenblatt, K.A.; Ray, R.M. Re: “Breastfeeding and reduced risk of breast cancer in an Icelandic cohort study”. Am. J. Epidemiol. 2001, 154, 975–977. [Google Scholar] [CrossRef]
- Russo, J.; Moral, R.; Balogh, G.A.; Mailo, D.; Russo, I.H. The protective role of pregnancy in breast cancer. Breast Cancer Res. 2005, 7, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Russo, J.; Russo, I.H. Role of differentiation in the pathogenesis and prevention of breast cancer. Endocr. Relat. Cancer 1997, 4, 7–21. [Google Scholar] [CrossRef]
- Srivastava, P.; Russo, J.; Russo, I.H. Chorionic gonadotropin inhibits rat mammary carcinogenesis through activation of programmed cell death. Carcinogenesis 1997, 18, 1799–1808. [Google Scholar] [CrossRef]
- Ginger, M.R.; Gonzalez-Rimbau, M.F.; Gay, J.P.; Rosen, J.M. Persistent changes in gene expression induced by estrogen and progesterone in the rat mammary gland. Mol. Endocrinol. 2001, 15, 1993–2009. [Google Scholar] [CrossRef]
- D'Cruz, C.M.; Moody, S.E.; Master, S.R.; Hartman, J.L.; Keiper, E.A.; Imielinski, M.B.; Cox, J.D.; Wang, J.Y.; Ha, S.I.; Keister, B.A.; et al. Persistent parity-induced changes in growth factors, TGF-beta3, and differentiation in the rodent mammary gland. Mol. Endocrinol. 2002, 16, 2034–2051. [Google Scholar] [CrossRef]
- Henry, M.D.; Triplett, A.A.; Oh, K.B.; Smith, G.H.; Wagner, K.U. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene 2004, 23, 6980–6985. [Google Scholar] [CrossRef]
- Medina, D. Breast cancer: The protective effect of pregnancy. Clin. Cancer Res. 2004, 10, 380S–384S. [Google Scholar] [CrossRef]
- Endocrine control of breast development. In Molecular Basis of Breast Cancer: Prevention and Treatment; Russo, J.; Rosen, I.H. (Eds.) Springer: Berlin, Germany, 2004; pp. 64–67.
- Ginger, M.R.; Rosen, J.M. Pregnancy-induced changes in cell-fate in the mammary gland. Breast Cancer Res. 2003, 5, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Misra, Y.; Bentley, P.A.; Bond, J.P.; Tighe, S.; Hunter, T.; Zhao, F.Q. Mammary gland morphological and gene expression changes underlying pregnancy protection of breast cancer tumorigenesis. Physiol. Genomics 2012, 44, 76–88. [Google Scholar] [CrossRef]
- Santucci-Pereira, J.; George, C.; Armiss, D.; Russo, I.; Vanegas, J.; Sheriff, F.; Lopez de Cicco, R.; Su, Y.; Russo, R.; Bidinotto, L.; et al. Mimicking pregnancy as a strategy for breast cancer prevention. Breast Cancer Manag. 2013, 2, 283–294. [Google Scholar]
- Belitskaya-Levy, I.; Zeleniuch-Jacquotte, A.; Russo, J.; Russo, I.H.; Bordas, P.; Ahman, J.; Afanasyeva, Y.; Johansson, R.; Lenner, P.; Li, X.; et al. Characterization of a genomic signature of pregnancy identified in the breast. Cancer Prev. Res. 2011, 4, 1457–1464. [Google Scholar] [CrossRef]
- Peri, S.; de Cicco, R.L.; Santucci-Pereira, J.; Slifker, M.; Ross, E.A.; Russo, I.H.; Russo, P.A.; Arslan, A.A.; Belitskaya-Levy, I.; Zeleniuch-Jacquotte, A.; et al. Defining the genomic signature of the parous breast. BMC Med. Genomics 2012, 5, 46–57. [Google Scholar] [CrossRef]
- Russo, J.; Rivera, R.; Russo, I.H. Influence of age and parity on the development of the human breast. Breast Cancer Res. Treat. 1992, 23, 211–218. [Google Scholar] [CrossRef]
- Russo, I.H.; Russo, J. Pregnancy-induced changes in breast cancer risk. J. Mammary Gland Biol. Neoplasia 2011, 16, 221–233. [Google Scholar] [CrossRef]
- Molecular Basis of Breast Cancer: Prevention and Treatment; Russo, J.; Russo, I.H. (Eds.) Springer-Verlag: Berlin, Germany, 2004; p. 447.
- Russo, J.; Santucci-Pereira, J.; de Cicco, R.L.; Sheriff, F.; Russo, P.A.; Peri, S.; Slifker, M.; Ross, E.; Mello, M.L.; Vidal, B.C.; et al. Pregnancy-induced chromatin remodeling in the breast of postmenopausal women. Int. J. Cancer 2012, 131, 1059–1070. [Google Scholar] [CrossRef]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef]
- Herrmann, A.; Fleischer, K.; Czajkowska, H.; Muller-Newen, G.; Becker, W. Characterization of cyclin L1 as an immobile component of the splicing factor compartment. FASEB J. 2007, 21, 3142–3152. [Google Scholar] [CrossRef]
- Russo, I.H.; Russo, J. Mammary gland neoplasia in long-term rodent studies. Environ. Health Perspect. 1996, 104, 938–967. [Google Scholar] [CrossRef]
- Russo, J.; Tait, L.; Russo, I.H. Susceptibility of the mammary gland to carcinogenesis. III. The cell of origin of rat mammary carcinoma. Am. J. Pathol. 1983, 113, 50–66. [Google Scholar]
- Tan, P.H.; Goh, B.B.; Chiang, G.; Bay, B.H. Correlation of nuclear morphometry with pathologic parameters in ductal carcinoma in situ of the breast. Mod. Pathol. 2001, 14, 937–941. [Google Scholar] [CrossRef]
- Bussolati, G.; Marchio, C.; Gaetano, L.; Lupo, R.; Sapino, A. Pleomorphism of the nuclear envelope in breast cancer: A new approach to an old problem. J. Cell Mol. Med. 2008, 12, 209–218. [Google Scholar]
- Palmer, J.E.; Sant Cassia, L.J.; Irwin, C.J.; Morris, A.G.; Rollason, T.P. The prognostic value of nuclear morphometric analysis in serous ovarian carcinoma. Int. J. Gynecol. Cancer 2008, 18, 692–701. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Kubicek, S.; Schotta, G.; Lachner, M.; Sengupta, R.; Kohlmaier, A.; Perez-Burgos, L.; Linderson, Y.; Martens, J.H.; O'Sullivan, R.J.; Fodor, B.D.; et al. The role of histone modifications in epigenetic transitions during normal and perturbed development. Ernst. Schering Res. Found. Workshop 2006, 57, 1–27. [Google Scholar] [CrossRef]
- Lin, W.; Dent, S.Y. Functions of histone-modifying enzymes in development. Curr. Opin. Genet. Dev. 2006, 16, 137–142. [Google Scholar] [CrossRef]
- Zuo, T.; Tycko, B.; Liu, T.M.; Lin, H.J.; Huang, T.H. Methods in DNA methylation profiling. Epigenomics 2009, 1, 331–345. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Down, T.A.; Thorne, N.P.; Flicek, P.; Kulesha, E.; Graf, S.; Tomazou, E.M.; Backdahl, L.; Johnson, N.; Herberth, M.; et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res. 2008, 18, 1518–1529. [Google Scholar] [CrossRef]
- Aiyar, S.E.; Cho, H.; Lee, J.; Li, R. Concerted transcriptional regulation by BRCA1 and COBRA1 in breast cancer cells. Int. J. Biol. Sci. 2007, 3, 486–492. [Google Scholar] [CrossRef]
- Ingenuity® Pathway Analysis. v.18030641. Available online: http://www.ingenuity.com/ (accessed on 24 January 2014).
- Habas, R.; Dawid, I.B. Dishevelled and Wnt signaling: Is the nucleus the final frontier? J. Biol. 2005, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Goode, E.L.; Fredericksen, Z.S.; Vierkant, R.A.; Pankratz, V.S.; Liu-Mares, W.; Rider, D.N.; Vachon, C.M.; Cerhan, J.R.; Olson, J.E.; et al. Association of genetic variation in genes implicated in the beta-catenin destruction complex with risk of breast cancer. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 2101–2108. [Google Scholar] [CrossRef]
- Ratcliffe, M.J.; Itoh, K.; Sokol, S.Y. A positive role for the PP2A catalytic subunit in Wnt signal transduction. J. Biol. Chem. 2000, 275, 35680–35683. [Google Scholar] [CrossRef]
- Cheyette, B.N.; Waxman, J.S.; Miller, J.R.; Takemaru, K.; Sheldahl, L.C.; Khlebtsova, N.; Fox, E.P.; Earnest, T.; Moon, R.T. Dapper, a Dishevelled-associated antagonist of beta-catenin and JNK signaling, is required for notochord formation. Dev. Cell 2002, 2, 449–461. [Google Scholar] [CrossRef]
- Zhao, L.; Hart, S.; Cheng, J.; Melenhorst, J.J.; Bierie, B.; Ernst, M.; Stewart, C.; Schaper, F.; Heinrich, P.C.; Ullrich, A.; et al. Mammary gland remodeling depends on gp130 signaling through Stat3 and MAPK. J. Biol. Chem. 2004, 279, 44093–44100. [Google Scholar]
- Chang, P.H.; Hwang-Verslues, W.W.; Chang, Y.C.; Chen, C.C.; Hsiao, M.; Jeng, Y.M.; Chang, K.J.; Lee, E.Y.; Shew, J.Y.; Lee, W.H. Activation of Robo1 signaling of breast cancer cells by Slit2 from stromal fibroblast restrains tumorigenesis via blocking PI3K/Akt/beta-catenin pathway. Cancer Res. 2012, 72, 4652–4661. [Google Scholar] [CrossRef]
- Bertucci, M.C.; Mitchell, C.A. Phosphoinositide 3-kinase and INPP4B in human breast cancer. Ann. N. Y. Acad. Sci. 2013, 1280, 1–5. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H. Three decades of Wnts: A personal perspective on how a scientific field developed. EMBO J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef]
- Turashvili, G.; Bouchal, J.; Burkadze, G.; Kolar, Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology 2006, 73, 213–223. [Google Scholar] [CrossRef]
- Vallorosi, C.J.; Day, K.C.; Zhao, X.; Rashid, M.G.; Rubin, M.A.; Johnson, K.R.; Wheelock, M.J.; Day, M.L. Truncation of the beta-catenin binding domain of E-cadherin precedes epithelial apoptosis during prostate and mammary involution. J. Biol. Chem. 2000, 275, 3328–3334. [Google Scholar] [CrossRef]
- Imbert, A.; Eelkema, R.; Jordan, S.; Feiner, H.; Cowin, P. Delta N89 beta-catenin induces precocious development, differentiation, and neoplasia in mammary gland. J. Cell Biol. 2001, 153, 555–568. [Google Scholar] [CrossRef]
- Howe, L.R.; Brown, A.M. Wnt signaling and breast cancer. Cancer Biol. Ther. 2004, 3, 36–41. [Google Scholar] [CrossRef]
- Meier-Abt, F.; Milani, E.; Roloff, T.; Brinkhaus, H.; Duss, S.; Meyer, D.S.; Klebba, I.; Balwierz, P.J.; van Nimwegen, E.; Bentires-Alj, M. Parity induces differentiation and reduces Wnt/Notch signaling ratio and proliferation potential of basal stem/progenitor cells isolated from mouse mammary epithelium. Breast Cancer Res. 2013, 15, R36. [Google Scholar] [CrossRef]
- Prasad, C.P.; Rath, G.; Mathur, S.; Bhatnagar, D.; Parshad, R.; Ralhan, R. Expression analysis of E-cadherin, Slug and GSK3beta in invasive ductal carcinoma of breast. BMC Cancer 2009, 9, 325–335. [Google Scholar] [CrossRef]
- Logullo, A.F.; Nonogaki, S.; Pasini, F.S.; Osorio, C.A.; Soares, F.A.; Brentani, M.M. Concomitant expression of epithelial-mesenchymal transition biomarkers in breast ductal carcinoma: Association with progression. Oncol. Rep. 2010, 23, 313–320. [Google Scholar]
- Kimelman, D.; Xu, W. Beta-catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef]
- Sugimura, R.; Li, L. Noncanonical Wnt signaling in vertebrate development, stem cells, and diseases. Birth Defects Res. C Embryo Today 2010, 90, 243–256. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Russo, J.; Santucci-Pereira, J.; Russo, I.H. The Genomic Signature of Breast Cancer Prevention. Genes 2014, 5, 65-83. https://doi.org/10.3390/genes5010065
Russo J, Santucci-Pereira J, Russo IH. The Genomic Signature of Breast Cancer Prevention. Genes. 2014; 5(1):65-83. https://doi.org/10.3390/genes5010065
Chicago/Turabian StyleRusso, Jose, Julia Santucci-Pereira, and Irma H. Russo. 2014. "The Genomic Signature of Breast Cancer Prevention" Genes 5, no. 1: 65-83. https://doi.org/10.3390/genes5010065
APA StyleRusso, J., Santucci-Pereira, J., & Russo, I. H. (2014). The Genomic Signature of Breast Cancer Prevention. Genes, 5(1), 65-83. https://doi.org/10.3390/genes5010065