2.1. The Theory
Assume that a sample of
n DNA sequences at a locus without recombination is taken from a single population evolving according to the Wright–Fisher model and all mutations are selectively neutral. The sample genealogy thus consists of 2(
n − 1) branches, each spanning at least one coalescent time (
Figure 1). The number of mutations that occurred in a branch is thus the sum of the numbers of mutations in the coalescent time it spans. Consider one branch, and without loss of generality, assume it spans the i-th coalescent time. Then, during the i-th coalescent time, the number of mutations occurred in the branch has expectation and variance equal to:
respectively. These are consequences of the coalescent time being exponentially distributed and the number of mutations in a given number of generations following a Poisson distribution. Consider the number of mutations in another branch that spans the j-th coalescent time. Then, the covariance between the two numbers of mutations is equal to:
if
i =
j, and zero otherwise.
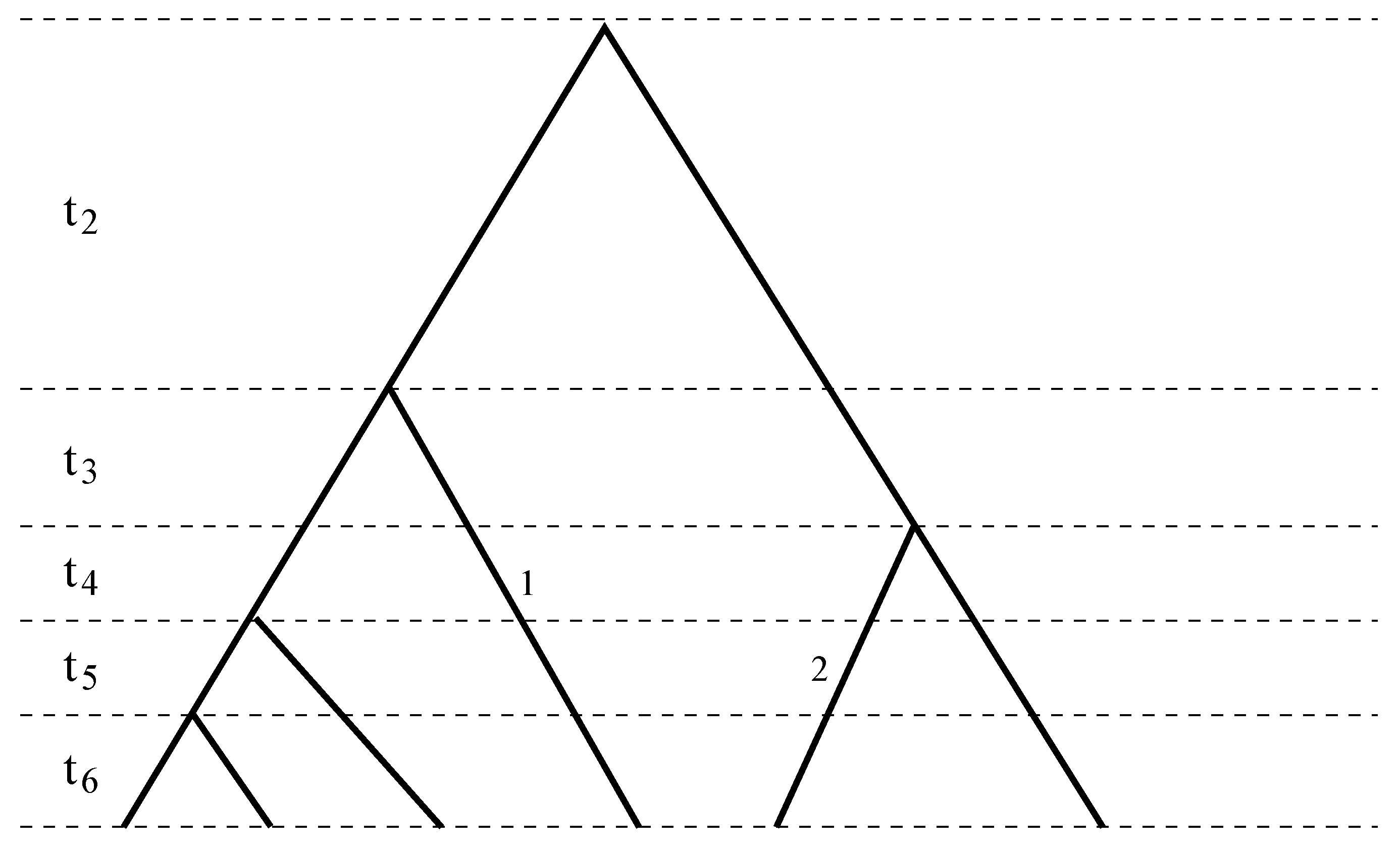
Figure 1.
A sample genealogy with different coalescent times separated by dashed lines. Branch 1 spans the third to the sixth coalescent times, χ1(2) = 0, χ1(i) = 1, for i = 3, ..., 6, while Branch 2 spans the fourth to the sixth coalescent times, χ2(2) = χ2(3) = 0, χ2(4) = χ2(5) = χ2(6) = 1. Combining Branches 1 and 2 results in ϕ(2) = 0, ϕ(3) = 1, ϕ(4) = ϕ(5) = ϕ(6) = 2.
Figure 1.
A sample genealogy with different coalescent times separated by dashed lines. Branch 1 spans the third to the sixth coalescent times, χ1(2) = 0, χ1(i) = 1, for i = 3, ..., 6, while Branch 2 spans the fourth to the sixth coalescent times, χ2(2) = χ2(3) = 0, χ2(4) = χ2(5) = χ2(6) = 1. Combining Branches 1 and 2 results in ϕ(2) = 0, ϕ(3) = 1, ϕ(4) = ϕ(5) = ϕ(6) = 2.
For the branch
k(
k = 1, ..., 2(
n − 1)) in the genealogy, define an index
χk(
i), such that it takes value one if the branch spans the
i-th coalescent time and zero otherwise. Then,
mk, the number of mutations, has its expectation and variance equal to:
respectively, and for two different branches
a and
b:
The previous results are readily generalized. Instead of considering the mutations in different branches separately, one can combine mutations in several branches. Suppose branches (
k1, ...,
kt) are combined. Define for the combined branches a variable
ϕ as:
Consider a population dynamics model in which the effective population size can change only at the time a coalescent event occurs. Although such a model does not stem from biological reality, its laddered changes in population sizes allow a reasonable approximation of reality and makes the mathematics simpler. Let
θi represent the
θ during the i-th coalescent period. Suppose the combined branches is denoted by branch (group)
k, then
mk, the number of mutations in branch k has expectation and variance equal to:
respectively, and for two such branches
a and
b, we have:
Suppose that the 2(
n − 1) branches of the sample genealogy are divided into
M (≤ 2(
n − 1)) disjoint groups (
i.e., each branch belongs to one and only one group). Let
mk represent the number of mutations in branch group
k and
m = (
m1, ...,
mk)
T. Then, similar to the previous result [
13], the relationship between
θ = (
θ2, ...,
θn)
T and
m can be expressed by a generalized linear model:
where
α is a matrix of dimension
M ×
n with:
and
e a vector of length
M representing error terms. Let Γ(
θ)=
Var(
m). Then:
where
γ and
β are both
M ×
M matrices defined as:
where
αk represents the k-th row vector of
α. Following the previous approach [
13,
14], a best linear unbiased estimator of
θ can be obtained as the limit of the series:
with
θ(0) being the initial estimate of
θ (for example, setting all
θi equal to Watterson’s estimate of
θ).
The above formulation allows maximal
n − 1 different values of
θ corresponding to the
n − 1 coalescent periods. Although very flexible, such an extreme model may lead to reduced accuracy of estimation for individual parameters, so some compromise is likely to be useful. When two or more consecutive
θ values are assumed to be the same, the length of the
θ vector will be reduced. At the extreme, if all of the
θs are the same,
θ is reduced to a single quantity, and when
M = 2 (
n − 1), it further defaults to BLUE [
13]. Since efficient estimators for a single value of
θ will continue to be useful in the analysis of the whole genome polymorphism of large samples, we will focus on developing one such scheme in this paper.
2.2. Allelic BLUE estimator
In order to take advantage of the BLUE estimator, sample genealogy needs to be inferred. Furthermore, the key to developing a highly efficient BLUE estimator is to define the M groups of branches, which not only retains the detail mutational information, but also satisfies the relationship
M << 2(
n − 1). Fu’s UPBLUE [
13] corresponds to the extreme in which
M = 2(
n − 1),
i.e., each branch belongs to its own group. While this may retain the maximal mutational information, it leads to computational inefficiency. Fu’s [
14] approach is more or less equivalent to
M =
n − 1, with groups defined by the size of mutations. This achieves computational efficiency with the expense of reduced accuracy due to over condensation of mutational information. Thus, the goal here is to strive for a balance.
We recognize that much of the phylogenetic information in a sample resides in the pattern of differences between distinct alleles. The phylogenetic method, UPGMA (e.g., [
16]), which was found to be appropriate in Fu [
13], will continue to be used in our new method. Since UPGMA is a sequential method, which at each step joins two sequences (or two groups of sequences) that differ the least. As a result, copies of sequences of the same allele will be joined together before any pair of sequences of distinct alleles. The resulting sample genealogy can be roughly divided into two portions (see
Figure 2), with the bottom portion corresponding to the coalescent within allelic class and the upper portion the coalescent among allelic classes. Combine all the branches (or segments of branches) underneath the dashed line into one group, which will be referred to as the within allele branch. Suppose there are
L distinct alleles in the sample; then we have for the within allele branch:
Then, the number of mutations in the within allele branch have expectation and variance equal respectively to:
Figure 2.
Schematic relationship between sample genealogy and allelic genealogy. The dark portion is the genealogy of distinct alleles, while the light portion (which is below the horizontal line) is the coalescent within alleles and contains no mutation or only a few in the dashed segments of the branches.
Figure 2.
Schematic relationship between sample genealogy and allelic genealogy. The dark portion is the genealogy of distinct alleles, while the light portion (which is below the horizontal line) is the coalescent within alleles and contains no mutation or only a few in the dashed segments of the branches.
Furthermore, we assume that there is no mutation in the within allele branch (which should be a good approximation, although technically, the assertion may not be true). Since the within allele branch does not span any coalescent time that overlaps with those of branches in the allelic genealogy, we have (assumed that the last branch group represents the within allele subtree) that:
where
α∗ a vector of length 2(
L − 1) with the
k-th element equal to
. The inverse of the covariance matrix of
m is:
where Γ
∗ is defined for branch groups of the allelic genealogy. Let m be the vector of mutations in branches of the allelic genealogy (the dark portion in the genealogy of
Figure 2). Then, Equation (15) becomes:
This limit will be referred to as the Allelic BLUE estimator denoted by θab. Since, for large samples, the number of distinct alleles is typically much smaller than the sample size; thus, the new estimator is expected to be highly efficient computationally.
To determine if it is indeed true that merging those branches representing within allele coalescent does not lead to significant loss of information and, thus, would not reduce the accuracy of estimation, we compared Allelic BLUE with the original BLUE using simulated samples for a number of combinations of θ and n. The correlation between the two estimates is around 0.99. Therefore, Allelic BLUE is expected to be as accurate as BLUE without merging branches.
2.3. Bias-Corrected Allelic BLUE Estimator
Since UPGMA systematically introduces bias in the inferred sample genealogy, the resulting Allelic BLUE estimate is expected to be biased similar to the BLUE estimator [
13]. Therefore, it is necessary to correct the bias. Similar to Fu [
13], we used simulated samples to derive understanding of the pattern of biases. A total of 550 combinations of
θ and
n were examined with 25 different
θ values: 0.5, 0.75, 1, 1.5, 2(1)5, 6(2)12, 15(5), 50, 60(10)100 and 150, and 25 different sample sizes
n: 10(5)25, 30(10)60, 80, 100(25)200, 250, 300, 400, 500, 750, 1000(1000)5000. For each combination of the parameters, 1000 samples were simulated, and for each simulated sample,
θab was obtained and their mean value computed over all simulated samples. Similar to those in Fu [
13], the estimates in almost all situations are underestimates of the true
θ. In general, the underestimate is the result of systematic bias of the UPGMA algorithm used to construct the genealogy, because UPGMA leads to early coalescent for more similar sequences and, thus, has a tendency to place more mutations in branches that are deeper into the tree. In the current situation, it is further compounded by our simplification that, up to the
i + 1 coalescent, there are no mutations.
Using regression analysis, Fu [
13] showed that the relationship:
summarizes well the BLUE estimate (with
M = 2(
n − 1)),
θ and sample size n, which is not larger than 100. When larger sample sizes were examined, the above equation is not adequate, and log transformation, rather than square-root transformation, can lead to a better regression [
17]. Therefore, log-transformation was chosen in our regression analysis.
Table 1 showed that
ln(
θab) can be summarized very well by the following equation:
which leads to an estimate of
θ from the solution for the above quadratic equation with regard to
ln(
θa):
where:
Although this estimator in most situations is excellent, we found that regression equations for a narrower range of sample sizes provides estimates that are more robust in some situations (particularly when
θ is large). As a result, we derive our final estimator
of
θ using Equation (23) with values of
a,
b and
c, as provided in
Table 2.
Table 1.
Summary of regression analysis between θab, θ and n.
Table 1.
Summary of regression analysis between θab, θ and n.
| Source | Sum of Squares | Degrees of freedom | Mean Square | |
|---|
| Model | 1,715.227 | 5 | 343.0454 | |
| Residual | 0.074 | 644 | 0.0001 | |
| Total | 1,715.301 | 649 | 2.6430 | |
| Term | Coefficient | Standard Error | ttest | P > |t| |
| ln(n) | 0.0080 | 0.0005 | 16.73 | 0.000 |
| ln(θ) | 1.0043 | 0.0025 | 398.16 | 0.000 |
| ln(n)ln(θ) | -0.0019 | 0.0004 | -4.19 | 0.000 |
| ln2(θ) | 0.0108 | 0.0005 | 19.80 | 0.000 |
| ln(n)ln2(θ) | -0.0012 | 0.0001 | -12.74 | 0.000 |
| constant | -0.1981 | 0.0027 | -72.99 | 0.000 |
Table 2.
Coefficients for estimating θ using Equation (23).
Table 2.
Coefficients for estimating θ using Equation (23).
| | Coefficients (
n’ = ln(n)) |
|---|
| Sample Size | a | b | c |
|---|
| n < 50 | 0.0112 − 0.0012
n’ | 1.0076 − 0.0026
n’ | −
ln(θa) − 0.2101 + 0.0107n’ |
| 50 ≤ n < 500 | 0.0131 − 0.0017
n’ | 1.0009 − 0.0016
n’ | −
ln(θa) − 0.1980 + 0.0088n’ |
| n ≥ 500 | 0.0069 − 0.0007
n’ | 0.9850 − 0.0008
n’ | −
ln(θa) − 0.1581 + 0.0025n’ |
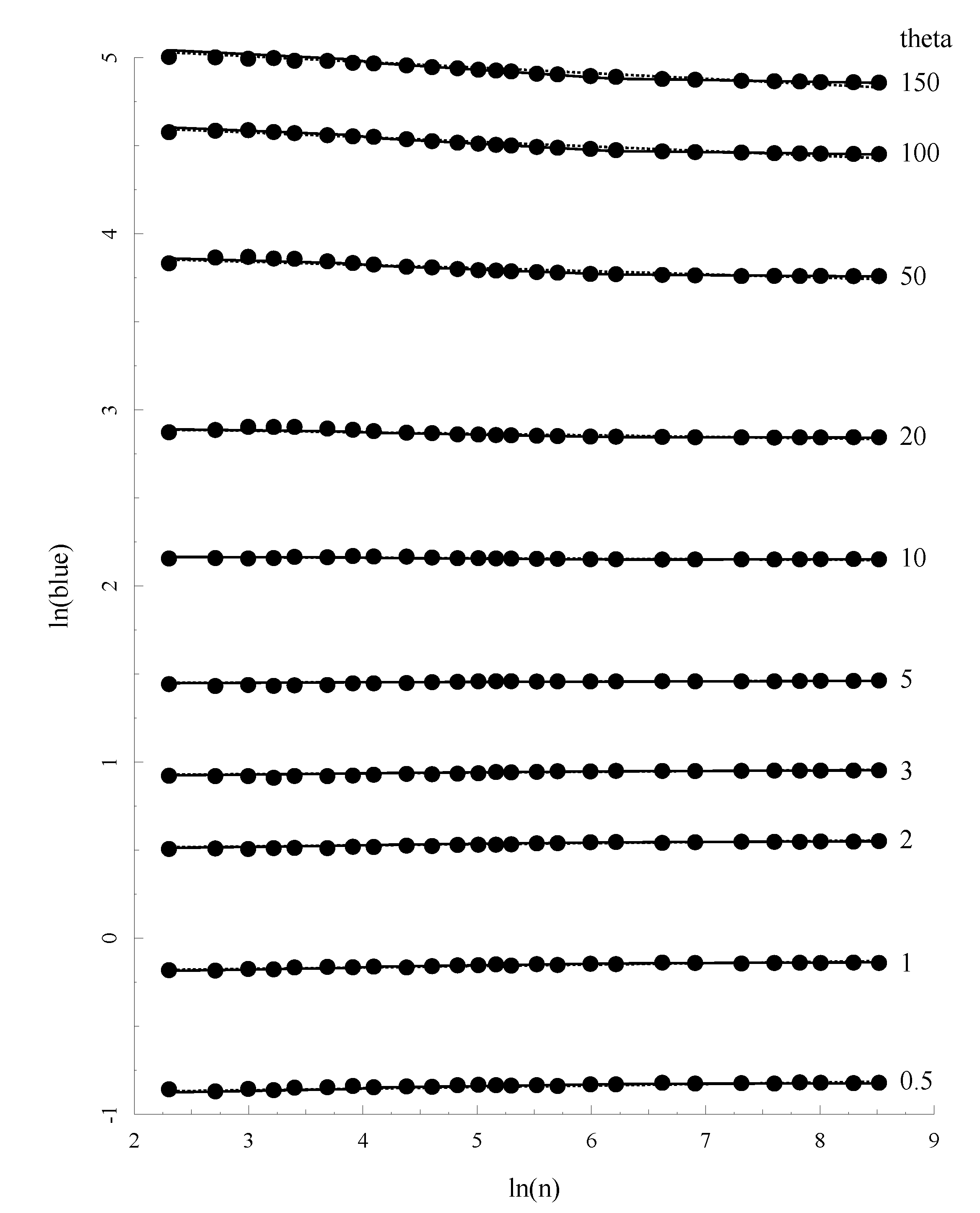
Figure 3 plots the relationship between
θ, sample size (
n) and the allelic BLUE estimates (
θab) for a subset of these parameter combinations. It is easy to see that the match between prediction and the mean value of
θa is excellent.
Figure 4 shows the distributions of the estimate
from simulated samples in the case of
n = 500 with
θ = 5, and
n = 2000 with
θ = 50, respectively. It appears in both cases that the normalities are sufficiently accurate approximations, which is indeed expected from the theory of least squares estimators.
The ultimate measure of the quality of an estimator is its bias and standard deviation for samples independent of those used to derive the estimator. Therefore, we simulated another set of samples for a number of combinations of
θ and
n and applied
, as well as UPBLUE to these samples.
Table 3 presents the summary of these simulations, particularly the efficiency of the new approach.
Figure 3.
The relationship between Allelic Blue estimate
θa,
θ and sample size. Solid lines represents the prediction of
θa based on Equation (23) with the coefficients given in
Table 2
Figure 3.
The relationship between Allelic Blue estimate
θa,
θ and sample size. Solid lines represents the prediction of
θa based on Equation (23) with the coefficients given in
Table 2
Figure 4.
Distribution of based on 1000 simulated samples. (Left) n = 500 and θ = 5; (right) n = 2000 and θ= 50.
Figure 4.
Distribution of based on 1000 simulated samples. (Left) n = 500 and θ = 5; (right) n = 2000 and θ= 50.
Table 3.
Performance of .
Table 3.
Performance of .
| θ | n | mean | SE | SDmin | Speed | Ratio |
|---|
| 2 | 20 | 1.97 | 1.00 | 0.97 | 0.00 | 10 |
| | 50 | 1.98 | 0.84 | 0.81 | 0.00 | 59 |
| | 100 | 1.98 | 0.75 | 0.74 | 0.00 | 280 |
| | 500 | 2.00 | 0.63 | 0.61 | 0.02 | 1,569 |
| | 1,000 | 2.00 | 0.58 | 0.58 | 0.27 | 1,615 |
| | 2,500 | 2.00 | 0.54 | 0.54 | 2.59 | 3,104 |
| 5 | 20 | 4.93 | 1.90 | 1.83 | 0.00 | 4.5 |
| | 50 | 4.96 | 1.52 | 1.48 | 0.00 | 23 |
| | 100 | 4.99 | 1.34 | 1.30 | 0.00 | 78 |
| | 500 | 5.01 | 1.08 | 1.05 | 0.04 | 970 |
| | 1,000 | 5.00 | 1.00 | 0.98 | 0.47 | 1,306 |
| | 2,500 | 4.91 | 0.87 | 0.90 | 4.90 | 1,565 |
| 20 | 20 | 20.33 | 5.84 | 5.52 | 0.00 | 2.0 |
| | 50 | 20.27 | 4.20 | 4.05 | 0.01 | 4.4 |
| | 100 | 20.06 | 3.47 | 3.37 | 0.06 | 8.7 |
| | 500 | 20.04 | 2.56 | 2.49 | 0.18 | 359 |
| | 1,000 | 19.99 | 2.32 | 2.26 | 0.91 | 727 |
| | 2,500 | 19.96 | 2.07 | 2.04 | 16 | 842 |
| 50 | 20 | 50.92 | 12.58 | 12.51 | 0.01 | 1.6 |
| | 50 | 50.45 | 8.68 | 8.59 | 0.02 | 2.5 |
| | 100 | 50.09 | 6.99 | 6.79 | 0.07 | 3.8 |
| | 500 | 50.06 | 4.70 | 4.58 | 0.97 | 72 |
| | 1,000 | 49.90 | 4.15 | 4.06 | 3.6 | 206 |
| | 2,500 | 49.80 | 3.94 | 3.57 | 42 | 356 |
| 100 | 20 | 100.25 | 23.47 | 24.03 | 0.01 | 1.5 |
| | 50 | 100.20 | 15.52 | 15.87 | 0.15 | 1.7 |
| | 100 | 99.97 | 12.24 | 12.08 | 0.20 | 2 |
| | 500 | 100.38 | 7.86 | 7.48 | 4 | 22 |
| | 1,000 | 99.89 | 6.59 | 6.47 | 16 | 49 |
| | 2,500 | 99.69 | 5.58 | 5.54 | 75 | 202 |
Table 3 shows that the speed of increases with the sample size slowly, while it increases faster with θ. This is because ’s speed is dependent on the number of alleles in the sample, which is more closely related to θ than sample size. In comparison, UPBLUE is considerably less efficient, particularly with increasing sample size. Take the case of θ = 100 and n = 5000, it takes on average about one minute for to complete the estimation, while it takes about 6 h for UPBLUE to do the same.




{kind=link}
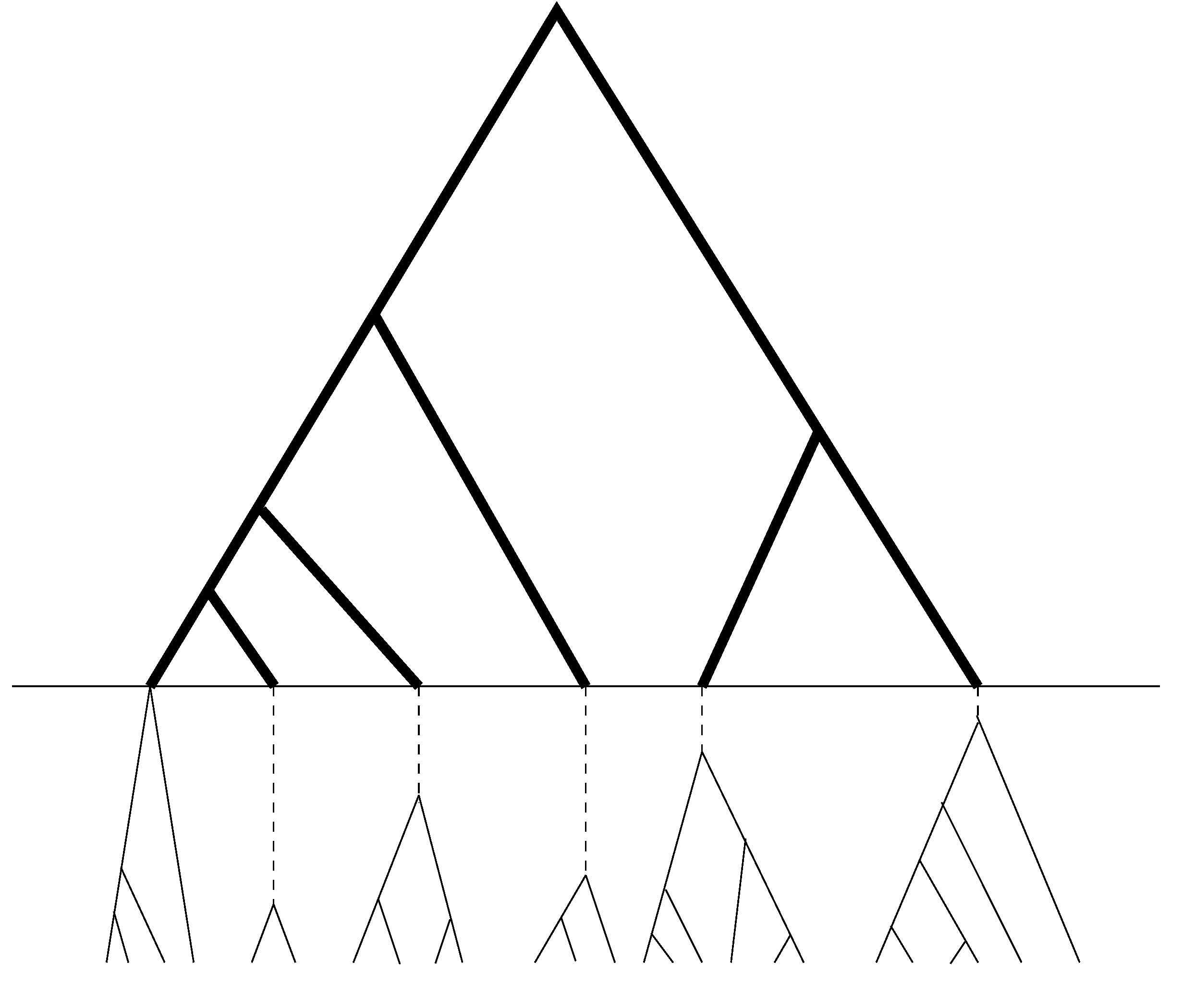{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



