Delivery of a Clinical Genomics Service

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
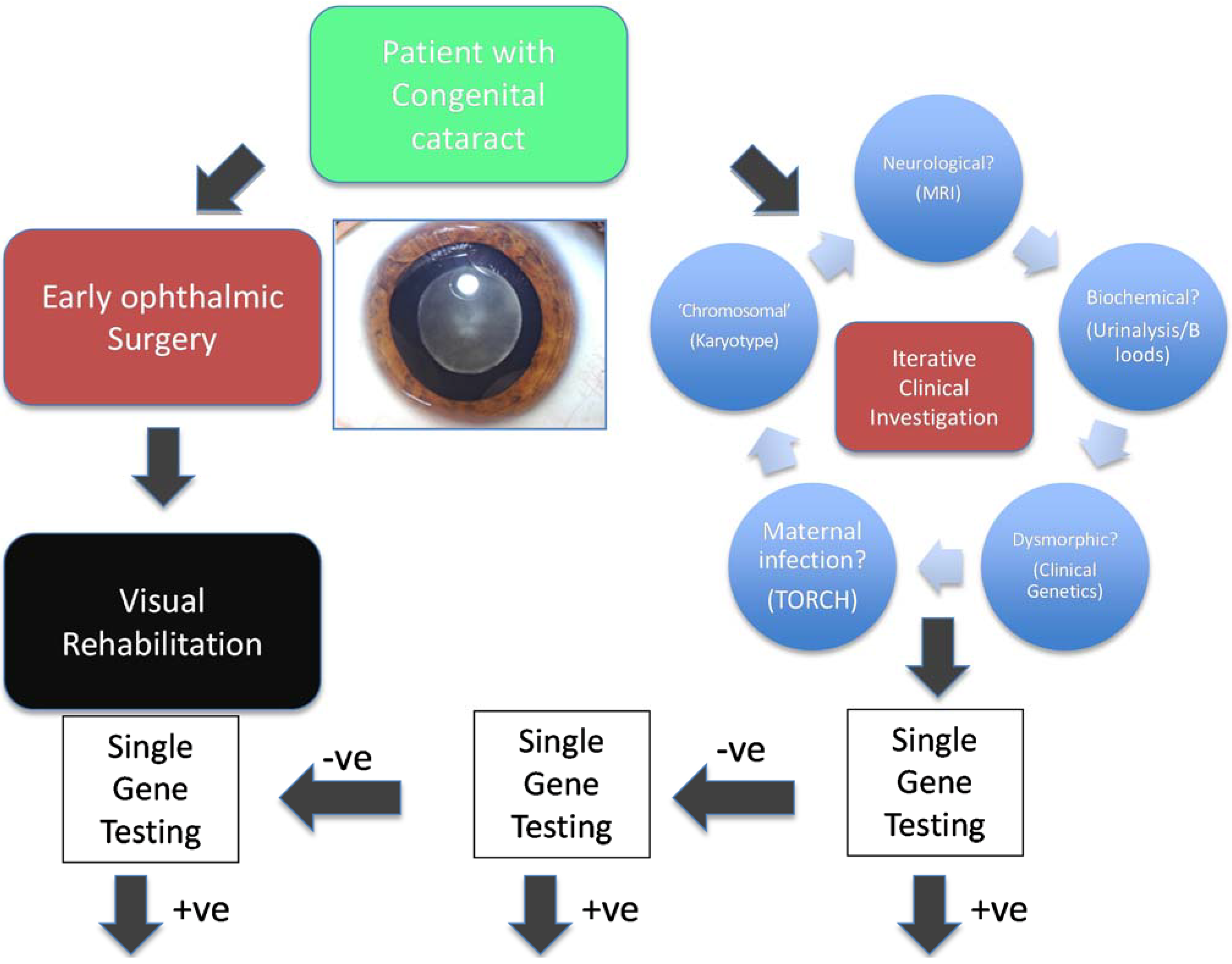
:1. Clinical Genetic Testing for Inherited Disorders
2. Next Generation Sequencing as a Diagnostic Tool


2.1. Targeted Next Generation Sequencing for Diagnostic Molecular Testing
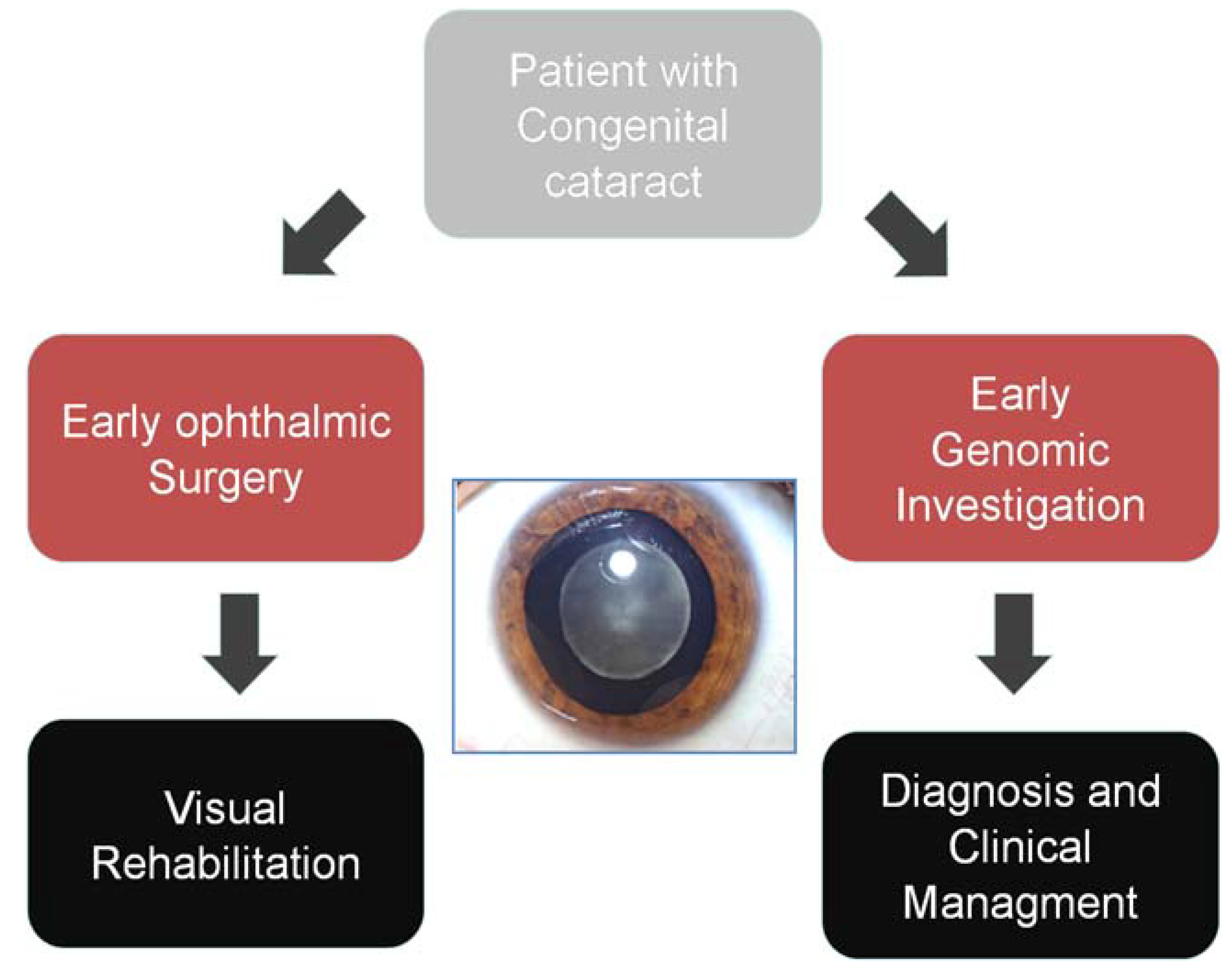
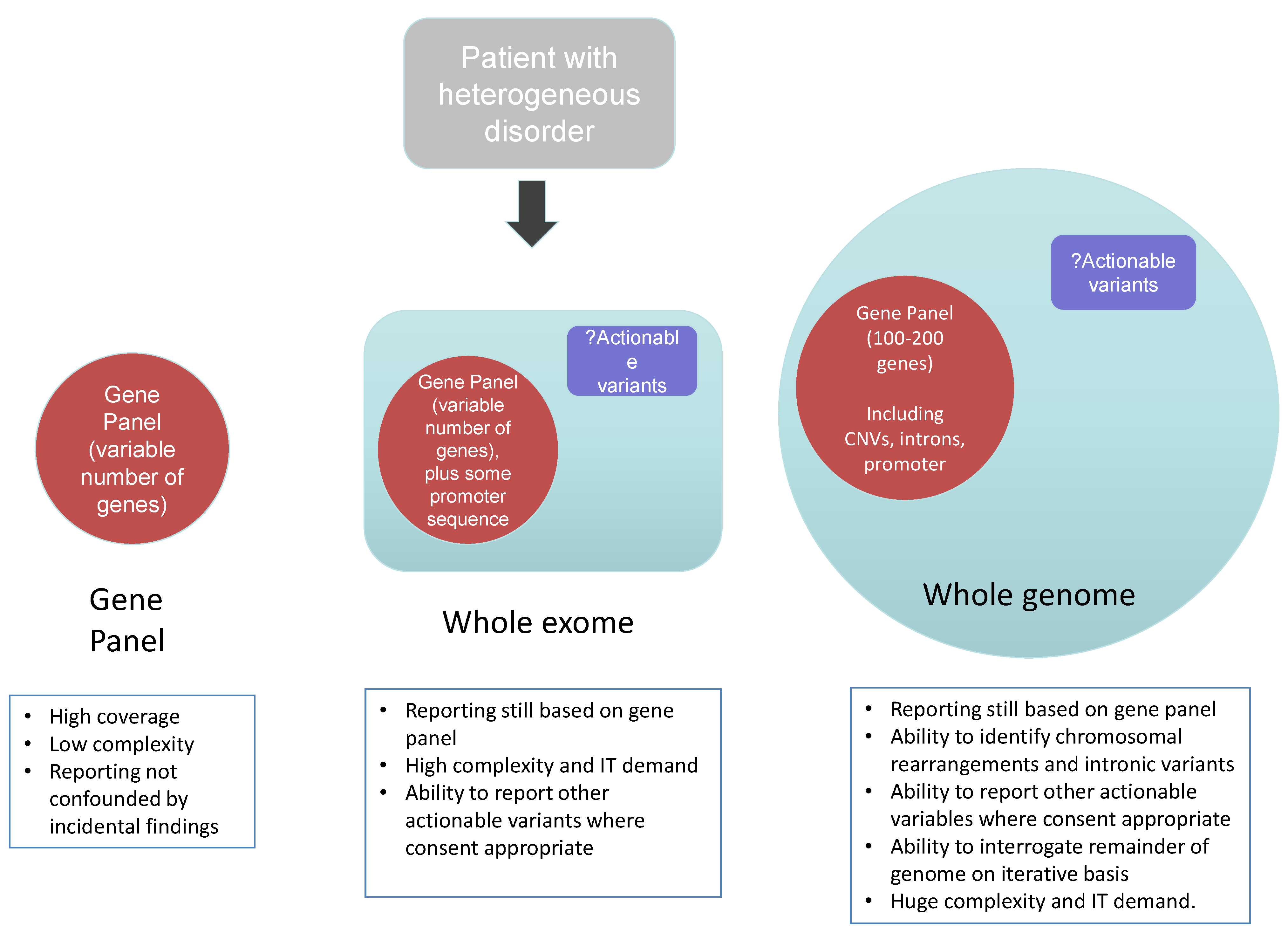
2.2. The Use of Genome-Wide NGS Approaches as a Diagnostic Tool

2.2.1. Clinical Exome Sequencing
- (i)
- Carrier status for a range of recessive disorders to inform future reproductive risks.
- (ii)
- Inherited disorders that are not predicted on the basis of family history or clinical presentation and for which treatment or preventive screening may be appropriate—so-called actionable variants. An example would be the detection of a variant in the low density lipoprotein receptor (LDLR) that would consistent with a diagnosis of familial hypercholesterolemia for which dietary intervention and statin treatment can reduce the risk of cardiovascular disease.
- (iii)
- Pharmacogenetic data that may reduce the risk of adverse drug reactions, e.g., detection of variants in thiopurine methyl transferase that predict adverse response to thiopurines, e.g., azathiopurine.
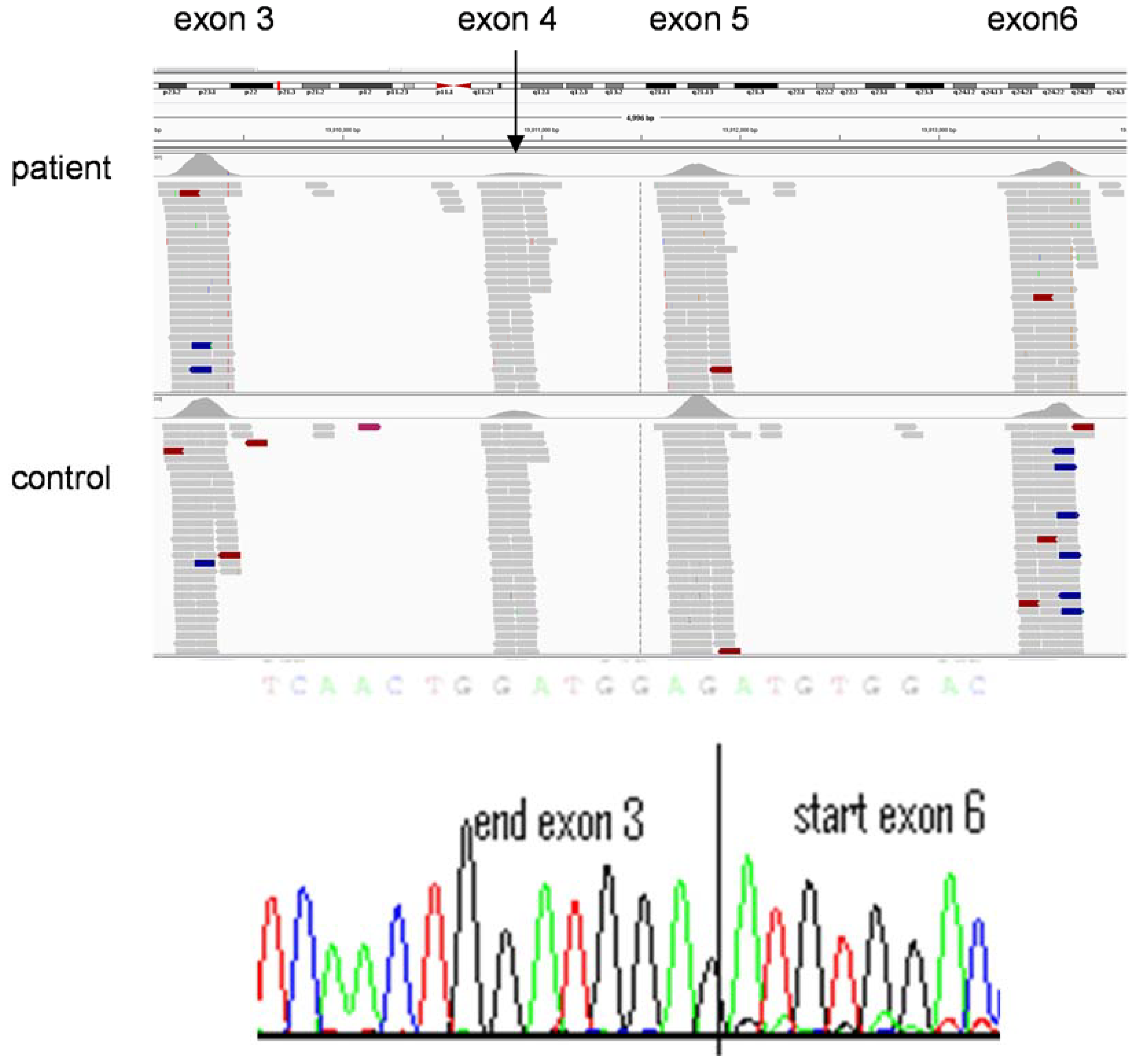
2.2.2. Whole Genome Sequencing
2.2.3. Methodological Considerations of Different NGS Approaches

3. Adoption of Clinical Genomics into Routine Clinical Practice
3.1. Training
3.2. Standardised Phenotyping
3.3. Ethical Issues
3.4. Economic and Societal Issues
4. Conclusions
Acknowledgements
Author Contributions
Conflict of Interests
References
- Chief Medical Officer. Rare is common. In Chief Medical Officer. Annual Report of the Chief Medical Officer 2009; Department of Health: London, UK, 2009; pp. 38–45. [Google Scholar]
- National Institutes of Health. Office of Rare Disease Research (ORDR). Available online: http://rarediseases.info.nih.gov/AboutUs.aspx (accessed on 10 August 2014).
- Genetic Testing Registry (GTR). Available online: http://www.ncbi.nlm.nih.gov/gtr/ (accessed on 10 August 2014).
- Macpherson, J.; Sawyer, H. Best practice guidelines for molecular diagnosis of Fragile X Syndrome. Available online: http://www.acgs.uk.com/quality-committee/best-practice-guidelines/ (accessed on 10 August 2014).
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G. Exome sequencing makes medical genomics a reality. Nat. Genet. 2010, 42, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L. Disease-targeted sequencing: A cornerstone in the clinic. Nat. Rev. Genet. 2013, 14, 295–300. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.; Mullaney, B.G.; Bhaskar, S.S.; Dickerson, J.E.; Hall, G.; O’Grady, A.; Webster, A.; Ramsden, S.C.; Black, G.C. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J. Med. Genet. 2012, 49, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hoischen, A.; Brunner, H.G.; Veltman, J.A. Disease gene identification strategies for exome sequencing. Eur. J. Hum. Genet. 2012, 20, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.A.; Mullins, R.F.; Wagner, A.H.; Andorf, J.L.; Johnston, R.M.; Bakall, B.B.; Deluca, A.P.; Fishman, G.A.; Lam, B.L.; Weleber, R.G.; et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet. 2013, 22, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Harismendy, O.; Ng, P.C.; Strausberg, R.L.; Wang, X.; Stockwell, T.B.; Beeson, K.Y.; Schork, N.J.; Murray, S.S.; Topol, E.J.; Levy, S.; et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009, 10. [Google Scholar] [CrossRef]
- Gillespie, R.L.; O’Sullivan, J.; Ashworth, J.; Bhaskar, S.; Williams, S.; Biswas, S.; Kehdi, E.; Ramsden, S.C.; Clayton-Smith, J.; Black, G.C.; et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology 2014. [Google Scholar] [CrossRef]
- Kocarnik, J.M.; Fullerton, S.M. Returning pleiotropic results from genetic testing to patients and research participants. JAMA 2014, 311, 795–796. [Google Scholar] [CrossRef] [PubMed]
- Black, G.C.; University of Manchester, Manchester, UK. Personal Communication, 2014.
- Clarke, A.J. Managing the ethical challenges of next-generation sequencing in genomic medicine. Br. Med. Bull. 2014, 111, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.A.; Rhodenizer, D.; da Silva, C.; Huff, I.J.; Keong, L.; Bean, L.J.; Coffee, B.; Collins, C.; Tanner, A.K.; He, M.; et al. Molecular diagnostic testing for congenital disorders of glycosylation (CDG): Detection rate for single gene testing and next generation sequencing panel testing. Mol. Genet. Metab. 2013, 110, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Manyika, J.; Chui, M.; Bughin, J.; Dobbs, R.; Bisson, P.; Marrs, A. Disruptive technologies: Advances that will transform life, business, and the global economy. McKinsey Global Institute. 2013. Available online: http://www.mckinsey.com/insights/business_technology/disruptive_technologies (accessed on 10 August 2014).
- Jacob, H.J. Next-generation sequencing for clinical diagnostics. N. Engl. J. Med. 2013, 369, 1557–1558. [Google Scholar] [CrossRef] [PubMed]
- Saunders, C.J.; Miller, N.A.; Soden, S.E.; Dinwiddie, D.L.; Noll, A.; Alnadi, N.A.; Andraws, N.; Patterson, M.L.; Krivohlavek, L.A.; Fellis, J.; et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Dixon-Salazar, T.J.; Silhavy, J.L.; Udpa, N.; Schroth, J.; Bielas, S.; Schaffer, A.E.; Olvera, J.; Bafna, V.; Zaki, M.S.; Abdel-Salam, G.H.; et al. Exome sequencing can improve diagnosis and alter patient management. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Worthey, E.A.; Mayer, A.N.; Syverson, G.D.; Helbling, D.; Bonacci, B.B.; Decker, B.; Serpe, J.M.; Dasu, T.; Tschannen, M.R.; Veith, R.L.; et al. Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet. Med. 2011, 13, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, M.N.; Wiszniewski, W.; Murdock, D.R.; Friedman, J.; Gonzaga-Jauregui, C.; Newsham, I.; Reid, J.G.; Fink, J.K.; Morgan, M.B.; Gingras, M.C.; et al. Whole-genome sequencing for optimized patient management. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Lupski, J.R.; Reid, J.G.; Gonzaga-Jauregui, C.; Rio Deiros, D.; Chen, D.C.; Nazareth, L.; Bainbridge, M.; Dinh, H.; Jing, C.; Wheeler, D.A.; et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N. Engl. J. Med. 2010, 362, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Shoemark, A.; Munye, M.M.; James, C.T.; Schmidts, M.; Patel, M.; Rosser, E.M.; Bacchelli, C.; Beales, P.L.; Scambler, P.J.; et al. Combined exome and whole-genome sequencing identifies mutations in ARMC4 as a cause of primary ciliary dyskinesia with defects in the outer dynein arm. J. Med. Genet. 2014, 51, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weedon, M.N.; Cebola, I.; Patch, A.M.; Flanagan, S.E.; de Franco, E.; Caswell, R.; Rodríguez-Seguí, S.A.; Shaw-Smith, C.; Cho, C.H.; Lango, A.H.; et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat. Genet. 2014, 46, 61–64. [Google Scholar] [CrossRef]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP). Available online: http://evs.gs.washington.edu/EVS/ (accessed on 10 August 2014).
- Gottlieb, B.; Beitel, L.K.; Trifiro, M. Changing genetic paradigms: Creating next-generation genetic databases as tools to understand the emerging complexities of genotype/phenotype relationships. Hum. Genomics 2014, 8. [Google Scholar] [CrossRef]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Torjesen, I. Genomes of 100,000 people will be sequenced to create an open access research resource. Br. Med. J. 2013, 347. [Google Scholar] [CrossRef]
- Köhler, S.; Doelken, S.C.; Mungall, C.J.; Bauer, S.; Firth, H.V.; Bailleul-Forestier, I.; Black, G.C.; Brown, D.L.; Brudno, M.; Campbell, J.; et al. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014, 42, D966–D974. [Google Scholar] [CrossRef]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Van El, C.G.; Cornel, M.C.; Borry, P.; Hastings, R.J.; Fellmann, F.; Hodgson, S.V.; Howard, H.C.; Cambon-Thomsen, A.; Knoppers, B.M.; Meijers-Heijboer, H.; et al. Whole-genome sequencing in health care: Recommendations of the European Society of Human Genetics. Eur. J. Hum. Genet. 2013, 21, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Lupski, J.R.; Biesecker, L.G. Reporting genomic sequencing results to ordering clinicians: Incidental, but not exceptional. JAMA 2013, 310, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.L.; Shih, V.E.; Madigan, P.M. Routine newborn screening for histidinemia. Clinical and biochemical results. N. Engl. J. Med. 1974, 291, 1214–1219. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.; McAllister, M.; Davies, L.M. Valuing the economic benefits of complex interventions: When maximising health is not sufficient. Health Econ. 2013, 22, 258–271. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newman, W.G.; Black, G.C. Delivery of a Clinical Genomics Service. Genes 2014, 5, 1001-1017. https://doi.org/10.3390/genes5041001
Newman WG, Black GC. Delivery of a Clinical Genomics Service. Genes. 2014; 5(4):1001-1017. https://doi.org/10.3390/genes5041001
Chicago/Turabian StyleNewman, William G., and Graeme C. Black. 2014. "Delivery of a Clinical Genomics Service" Genes 5, no. 4: 1001-1017. https://doi.org/10.3390/genes5041001
APA StyleNewman, W. G., & Black, G. C. (2014). Delivery of a Clinical Genomics Service. Genes, 5(4), 1001-1017. https://doi.org/10.3390/genes5041001