
PHF6 Degrees of Separation: The Multifaceted Roles of a Chromatin Adaptor Protein

Abstract
:1. Introduction
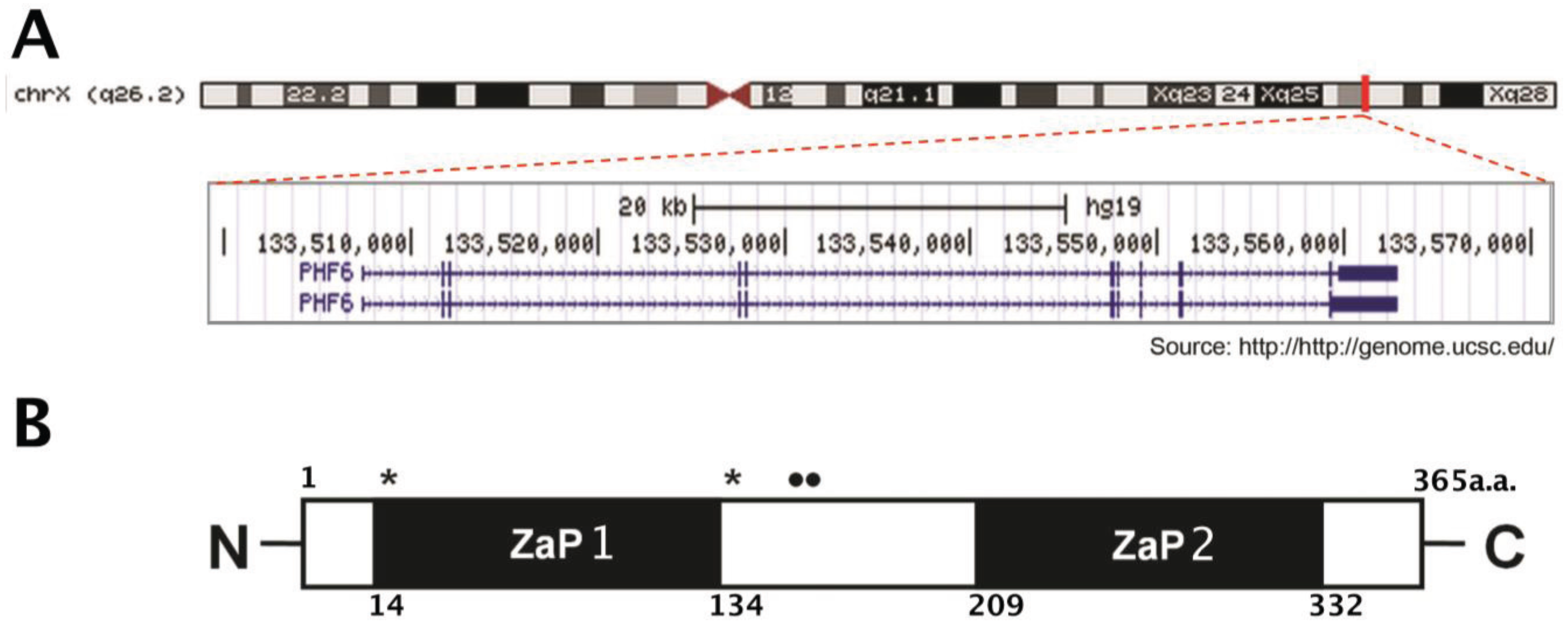
1.1. Structure and Expression Pattern of PHF6

1.2. Regulation of PHF6 Transcripts
2. Consequences of Germline Mutations of PHF6
2.1. Börjeson–Forssman–Lehmann Syndrome (BFLS)
2.2. Coffin–Siris syndrome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender | Nucleotide Change | Amino Acid Change | Type of Mutation | Location of Mutation | Cancer | Isolated3/ De novo | Reference |
|---|---|---|---|---|---|---|---|
| M | c.2T>C | p.M1T | Missense | Exon 2 | [9] | ||
| M | c.2T>C | p.M1T | Missense | Exon 2 | [32] | ||
| M | c.134G>A | p.C45Y | Missense | Exon 2 | [9] | ||
| M | c.134G>A | p.C45Y | Missense | Exon 2 | Isolated | [9] | |
| M | c.266G>T | p.G89V | Missense | Exon 4 | [45] | ||
| M | c.296G>T | p.C99F | Missense | Exon 4 | Isolated | [9] | |
| M, F | c.686A>G | p.H229R | Missense | Exon 7 | [9] | ||
| M | c.700A>G | p.K234E | Missense | Exon 7 | [9] | ||
| M | c.769A>G | p.R257G | Missense | Exon 8 | [9] | ||
| M | c.769A>G | p.R257G | Missense | Exon 8 | Yes2 | Isolated | [16] |
| M | c.940A>G | p.I314V | Missense | Exon 9 | [32] | ||
| M | c.22A>T | p.K8* | Nonsense | Exon 2 | [9] | ||
| F | c.955C>T | p.R319* | Nonsense | Exon 9 | De novo | [42] | |
| M | c.1024C>T | p.R342* | Nonsense | Exon 10 | De novo | [26,28] | |
| M | c.1024C>T | p.R342* | Nonsense | Exon 10 | [9] | ||
| M | c.1024C>T | p.R342* | Nonsense | Exon 10 | [9] | ||
| M | c.1024C>T | p.R342* | Nonsense | Exon 10 | [9] | ||
| M | c.1024C>T | p.R342* | Nonsense | Exon 10 | Yes3 | [31] | |
| F | c.27dupA | p.G10fs*21 | Frameshift | Exon 2 | De novo | [32] | |
| M | IVS2–8A>G | M46fsΔexon3 | Frameshift | Exon 3 | [16] | ||
| F | c.677delG | p.G226fsE*53 | Frameshift | Exon 7 | De novo | [41] | |
| F | c.914G>T | p.C305F | Frameshift | Exon 9 | De novo | [41] | |
| F | Duplication | Exons 4–5 | De novo | [42] | |||
| F | Duplication | Exons 4–5 | De novo | [42] | |||
| F | 6 kb deletion | Deletion | Exons 4–5 | De novo | [42] | ||
| F | 100 kb deletion | Deletion | Exons 6–10 | De novo | [44] | ||
| F | 15 kb deletion | Deletion | Exons 9–11 | De novo | [43] | ||
| M | c.999–1001 delTGA | p.D333del | Deletion | Exon 10 | [46] | ||
| M | c.999–1001 delTGA | p.D333del | Deletion | Exon 10 | [26,28] | ||
| F | Entire gene deleted | Deletion | Whole gene | De novo | [42] | ||
| F | 270 kb deletion | Deletion | Whole gene | Isolated | [44] |
3. Consequences of Somatic Mutations of PHF6
3.1. T-Cell Acute Lymphoblastic Leukemia (T-ALL)
3.2. Acute Myeloid Leukemia (AML)
3.3. PHF6 Loss-of-Function in Other Neoplasias
| Gender | Nucleotide Change | Amino Acid Change | Type of Mutation | Location of Mutation | Cancer1 | Reference |
|---|---|---|---|---|---|---|
| F | c.90_91insCCCG | p.L31PfsX6 | Insertion/Deletion | Exon 2 | T-ALL | [49] |
| M | p.G10fs | Frameshift | Exon 2 | T-ALL | [10] | |
| M | p.A41fs | Frameshift | Exon 2 | T-ALL | [10] | |
| M | p.H44fs | Frameshift | Exon 2 | T-ALL | [10] | |
| F | c.76-95del20+insTTGG | p.P26fs | Frameshift | Exon 2 | T-ALL | [48] |
| M | p.Y105fs | Frameshift | Exon 3 | T-ALL | [10] | |
| M | c.267_268insTTAGGACC | p.A90LfsX10 | Insertion/Deletion | Exon 4 | T-ALL | [49] |
| M | p.G122X | Nonsense | Exon 4 | T-ALL | [10] | |
| M | p.R116X | Nonsense | Exon 4 | T-ALL | [10] | |
| M | p.T98fs | Frameshift | Exon 4 | T-ALL | [10] | |
| F | c.289A>T | p.K97X | Nonsense | Exon 4 | T-ALL | [48] |
| M | p.H135fs | Frameshift | Exon 5 | T-ALL | [10] | |
| F | p.F172fs | Frameshift | Exon 6 | T-ALL | [10] | |
| M | p.R225X | Nonsense | Exon 6 | T-ALL | [10] | |
| M | p.S158fs | Frameshift | Exon 6 | T-ALL | [10] | |
| M | p.S191fs | Frameshift | Exon 6 | T-ALL | [10] | |
| M | c.525_526delGT | p.S176fs | Frameshift | Exon 6 | T-ALL | [48] |
| M | c.673C > T | p.R225X | Nonsense | Exon 7 | T-ALL | [49] |
| M | c.653_667delGGGAGGAAGAAAATGinsCCCTTTAAAGGGA | p.G218AfsX | Insertion/Deletion | Exon 7 | T-ALL | [49] |
| M | p.K235X | Nonsense | Exon 7 | T-ALL | [10] | |
| M | p.C215Y | Missense | Exon 7 | T-ALL | [10] | |
| M | p.G263fs | Frameshift | Exon 8 | T-ALL | [10] | |
| M | c.735M736dupTT | p.S246FfsX34 | Duplication | Exon 8 | T-ALL | [49] |
| M | p.K273X | Nonsense | Exon 8 | T-ALL | [10] | |
| M | p.C280Y | Missense | Exon 8 | T-ALL | [10] | |
| M | p.R257X | Nonsense | Exon 8 | T-ALL | [10] | |
| M | c.820T>C | p.R274X | Nonsense | Exon 8 | T-ALL | [10] |
| M | c.808C>T | p.Q270X | Nonsense | Exon 8 | T-ALL | [48] |
| M | c.823G>A | p.G275R | Missense | Exon 8 | T-ALL | [48] |
| M | c.779insCGGGAGGATCC | p.D262fs | Frameshift | Exon 8 | T-ALL | [48] |
| M | p.S320X | Nonsense | Exon 9 | T-ALL | [10] | |
| M | c.903C>A | p.Y301X | Nonsense | Exon 9 | T-ALL | [49] |
| M | p.Y303fs | Frameshift | Exon 9 | T-ALL | [10] | |
| M | p.C283R | Missense | Exon 9 | T-ALL | [10] | |
| M | p.T300A | Missense | Exon 9 | T-ALL | [10] | |
| M | p.A311P | Missense | Exon 9 | T-ALL | [10] | |
| M | p.Y303X | Nonsense | Exon 9 | T-ALL | [10] | |
| M | c.933_934insT | p.A311X | Nonsense | Exon 9 | T-ALL | [48] |
| M | c.835delA | p.K279fs | Frameshift | Exon 9 | T-ALL | [48] |
| M | c.1024C>T | p.R342* | Nonsense | Exon 10 | T-ALL | [31] |
| M | p.D333fs | Frameshift | Exon 10 | T-ALL | [10] | |
| M | c.986A>G | p.H329R | Missense | Exon 10 | T-ALL | [10] |
| M | c.973T>C | p.Y325H | Missense | Exon 10 | T-ALL | [48] |
| M | p.C215Y | Missense | Exon 7 | T-ALL | [10] | |
| M | p.C28fs | Frameshift | Exon 2 | T-ALL | [10] | |
| F | c.968 + 1G > A | Undetermined | Non-coding | Intron 9 | T-ALL | [49] |
| M | c.968+2T_968+5GdelTAAG | Undetermined | Non-coding | Intron 9 | T-ALL | [49] |
| M | 0.55 Mb deleted | Absent | Deletion | n/a | T-ALL | [10] |
| M | 0.23 Mb deleted | Absent | Deletion | n/a | T-ALL | [10] |
| M | 1.50 Mb deleted | Absent | Deletion | n/a | T-ALL | [10] |
| M | 0.27 Mb deleted | Absent | Deletion | n/a | T-ALL | [10] |
| M | 1.90 Mb deleted | Absent | Deletion | n/a | T-ALL | [10] |
| M | 0.20 Mb deleted | Absent | Deletion | n/a | T-ALL | [10] |
| M | 0.08 Mb deleted | Absent | Deletion | n/a | T-ALL | [10] |
| M | 0.11 Mb deleted | Absent | Deletion | n/a | T-ALL | [10] |
| M | p.C20fs | Frameshift | Exon 2 | AML | [11] | |
| M | p.A40G | Missense | Exon 2 | AML | [11] | |
| F | p.N171fs | Frameshift | Exon 6 | AML | [11] | |
| M | p.P200fs | Frameshift | Exon 7 | AML | [11] | |
| M | p.R274X | Nonsense | Exon 8 | AML | [11] | |
| M | p.R335fs | Frameshift | Exon 9 | AML | [11] | |
| M | p.H302Y | Missense | Exon 9 | AML | [11] | |
| M | p.R319X | Nonsense | Exon 9 | AML | [11] | |
| M | p.H329L | Missense | Exon 10 | AML | [11] | |
| M | p.R342X | Nonsense | Exon 10 | AML | [11] | |
| M | c.27dupA | p.K9RfsX12 | Duplication | Exon 2 | AML | [49] |
| M | c.83_101delGTGGACAGTTACTAATATCinsAT | P.C28YfsX2 | Insertion/Deletion | Exon 2 | AML | [49] |
| M | c.769A>G | p.R257G | Missense | Exon 8 | HA | [16] |
| M | c.673C > T | p.R225X | Nonsense | Exon 7 | HC | [9] |
| M | c.665C>T | p.A135V | Missense | Exon 5 | CML | [57] |
| M | c.670–679del10 | p.N137_E139del140fsX142 | Frameshift | Exon 5 | CML | [57] |
| M | c.895–896delTG | p.C212WfsX222 | Frameshift | Exon 7 | CML | [57] |
4. Delineating the Functional Interactions of PHF6
4.1. Functional Analysis of the Conserved Motifs within PHF6
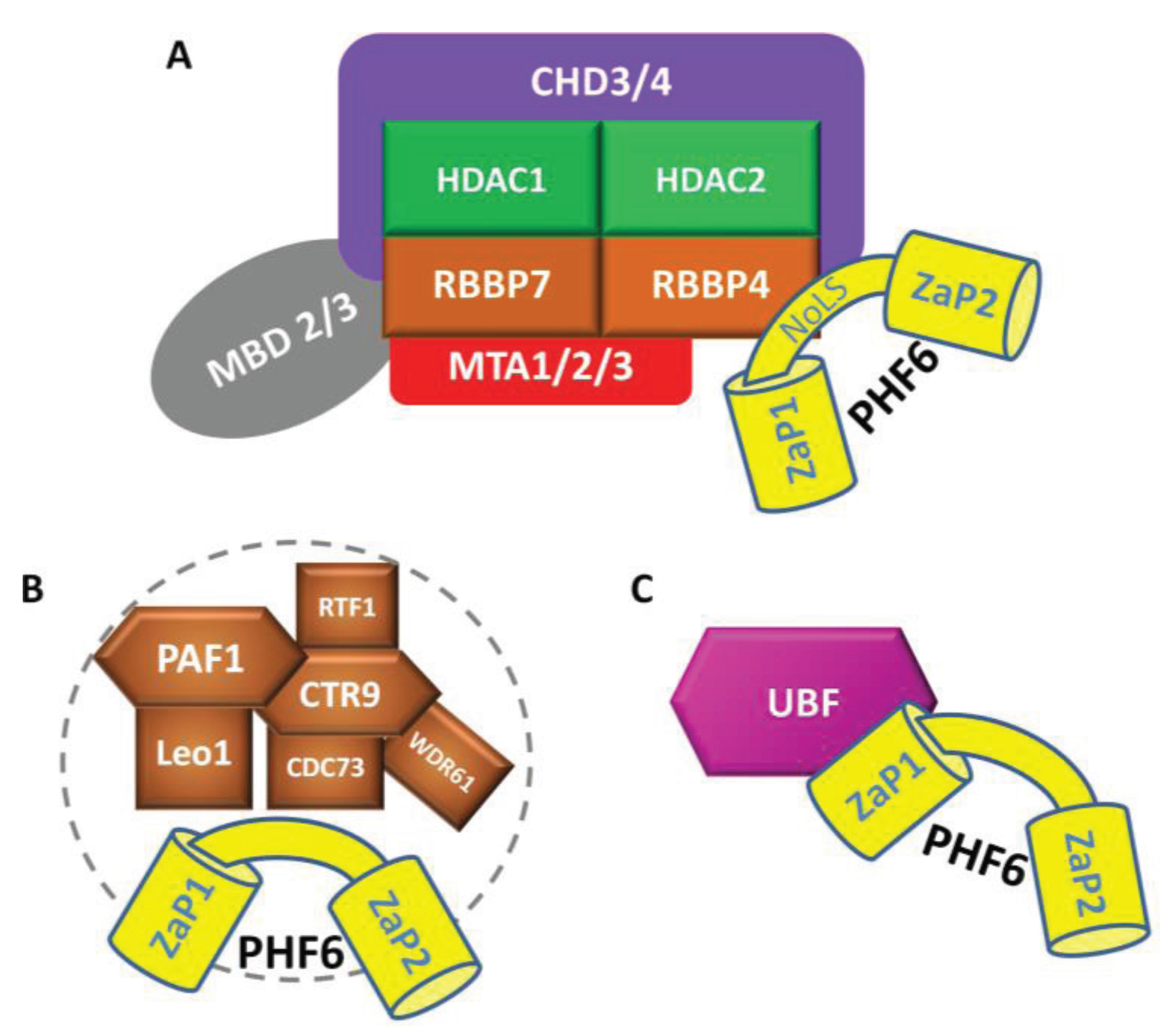
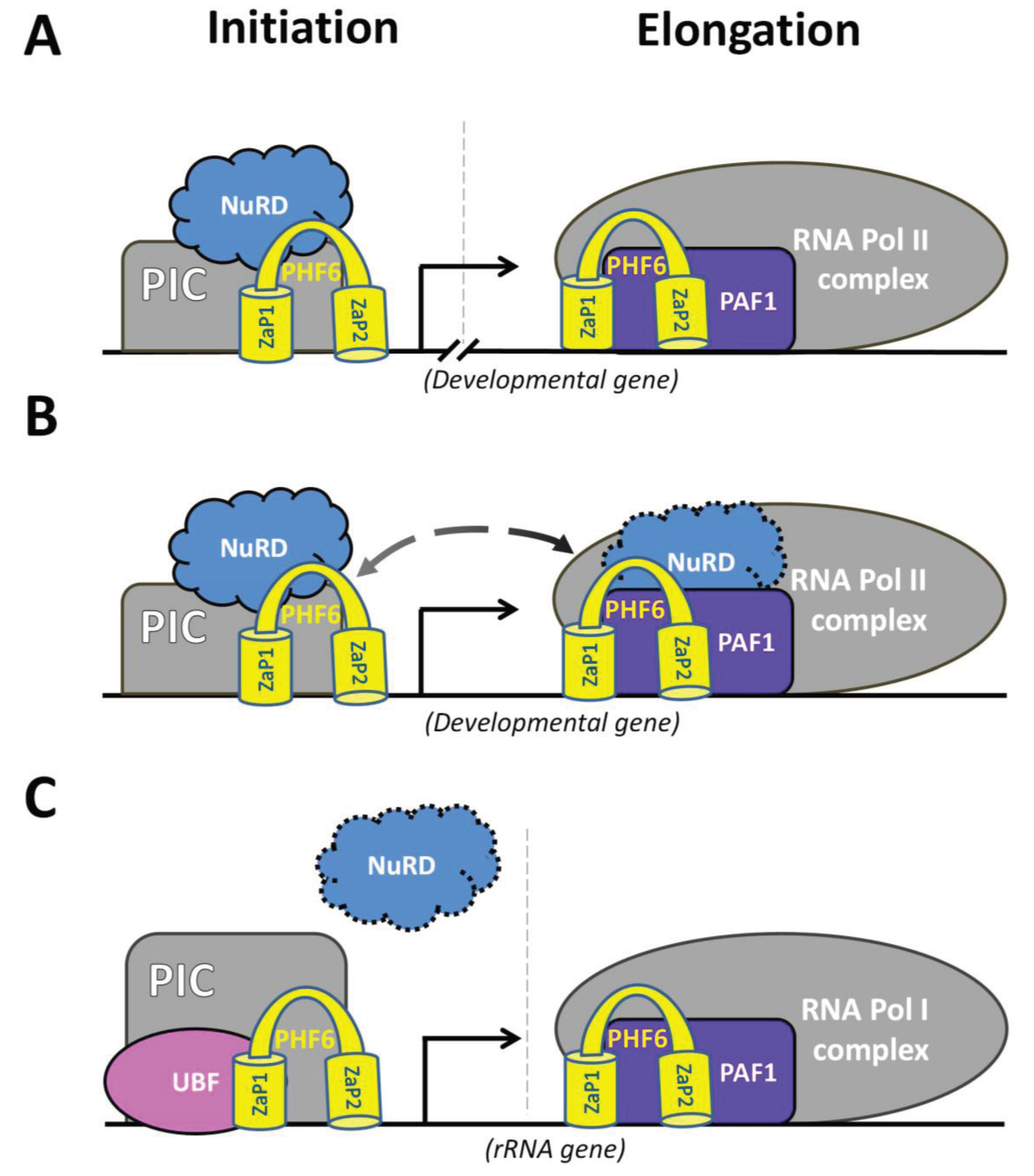
4.2. PHF6 Interacting Partners
4.2.1 Nucleosome Remodelling and Deacetylation (NuRD) complex
| Protein | Uniprot Accession # | Function | PZP Motif or ZaP Domain | Amino Acid Residues | Reference |
|---|---|---|---|---|---|
| PHF6 | Q8IWS0 | Transcriptional regulation | ZaP (x2) | 14–134, 209–332 | [14,87] |
| BRPF1 | P55201 | MOZ/MORF-dependent H3 acetylation | PZPM | 273–450 | [88] |
| BRPF2 | O95696 | MOZ/MORF-dependent H3 acetylation | PZPM | 214–391 | [88] |
| BRPF3 | Q9ULD4 | MOZ/MORF-dependent H3 acetylation | PZPM | 188–389 | [88] |
| G2E3 | Q7L622 | E3 ubiquitin-protein ligase | ZaP | 11–130 | [89] |
| JADE-1 | Q6IE81 | HBO1-dependent H4 acetylation | PZPM | 203–371 | [88] |
| JADE-2 | Q9NQC1 | HBO1-dependent H4 acetylation | PZPM | 199–367 | [88] |
| JADE-3 | Q92613 | HBO1-dependent H4 acetylation | PZPM | 200–368 | [88] |
| JMJD2A (KDM4A) | O75164 | H3K9/H3K36 demethylase | PZPM | 665–887 | [90] |
| JMJD2B (KDM4B) | O94953 | H3K9 demethylase | PZPM | 681–909 | [90] |
| JMJD2C (KDM4C) | Q9H3R0 | H3K9/H3K36 demethylase | PZPM | 642–867 | [90] |
| MLL1 (KMT2A) | Q03164 | H3K4 methyltransferase | PZPM | 1566–1980 | [91] |
| MLL2 (KMT2D) | O14686 | H3K4 methyltransferase | ZaP (x2) | 63–220, 5029–5139 | [92] |
| MLL3 (KMT2C) | Q8NEZ4 | H3K4 methyltransferase | ZaP (x2) | 131–333, 4399–4509 | [93] |
| MLL4 (KMT2B) | Q9UMN6 | H3K4 methyltransferase | PZPM | 1335–1688 | [93] |
| PHF7 | Q9BWX1 | Binds chromatin | ZaP | 30–147 | [94] |
| PHF11 | Q9UIL8 | Transcriptional regulation | ZaP | 42–162 | [95] |
| PHF14 | O94880 | Transcriptional regulation | PZPM | 319–501 | [96] |
| RAI1 | Q7Z5J4 | Transcriptional regulation | ZaP | 1780–1905 | [97] |
| TCF20 | Q9UGU0 | Transcriptional regulation | ZaP | 1789–1935 | [98] |
4.2.2 RNA Polymerase II Associated Factor 1 (PAF1)
4.2.3 Additional Interactions

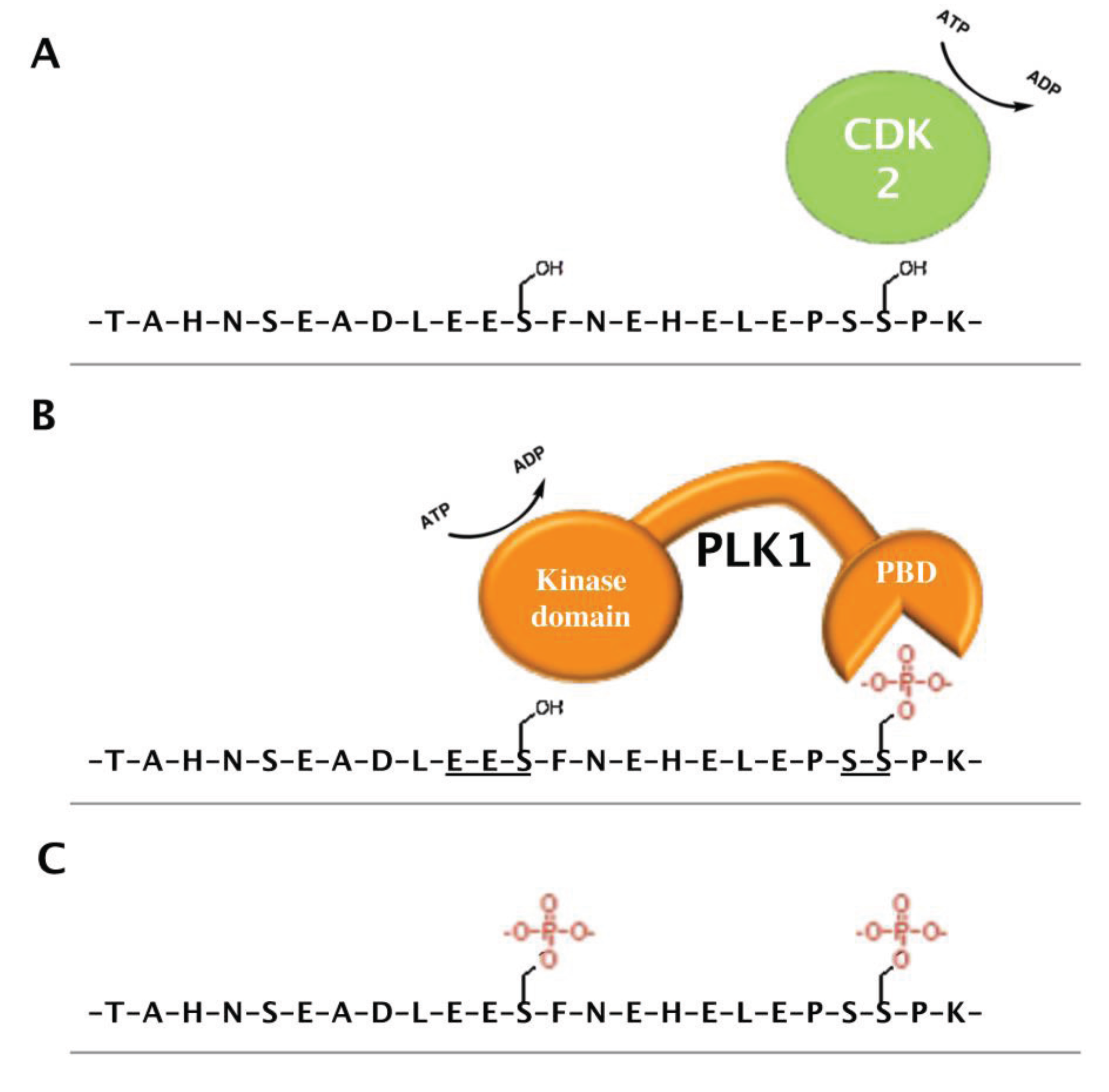
4.3. PHF6 Is a Putative Phosphoprotein

5. Perspectives and Future Directions
5.1. Predicting Clinical Outcomes for Patients Expressing PHF6 Loss-of-Function
5.2. Developmental Role of PHF6

5.3 Nucleolar Role of PHF6
6. Conclusion
Acknowledgements
Author Contributions
Conflicts of Interest
References and Notes
- van Bokhoven, H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 2011, 45, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies - the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone h3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Buchanan, K.L.; Tsien, F.; Jiang, G.; Sun, B.; Uicker, W.; Weemaes, C.M.; Smeets, D.; Sperling, K.; Belohradsky, B.H.; et al. DNA methyltransferase 3b mutations linked to the icf syndrome cause dysregulation of lymphogenesis genes. Hum. Mol. Genet. 2001, 10, 2917–2931. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Lower, K.M.; Turner, G.; Kerr, B.A.; Mathews, K.D.; Shaw, M.A.; Gedeon, A.K.; Schelley, S.; Hoyme, H.E.; White, S.M.; Delatycki, M.B.; et al. Mutations in phf6 are associated with borjeson-forssman-lehmann syndrome. Nat. Genet. 2002, 32, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Palomero, T.; Khiabanian, H.; Van der Meulen, J.; Castillo, M.; Van Roy, N.; De Moerloose, B.; Philippe, J.; Gonzalez-Garcia, S.; Toribio, M.L.; et al. Phf6 mutations in t-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Patel, J.; Abdel-Wahab, O.; Lobry, C.; Hedvat, C.V.; Balbin, M.; Nicolas, C.; Payer, A.R.; Fernandez, H.F.; Tallman, M.S.; et al. Phf6 mutations in adult acute myeloid leukemia. Leukemia 2011, 25, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, D.S.; Wagner, L.; Feingold, E.A.; Shenmen, C.M.; Grouse, L.H.; Schuler, G.; Klein, S.L.; Old, S.; Rasooly, R.; Good, P.; et al. The status, quality, and expansion of the nih full-length cdna project: The mammalian gene collection (mgc). Genome Res. 2004, 14, 2121–2127. [Google Scholar] [PubMed]
- Perry, J. The epc-n domain: A predicted protein-protein interaction domain found in select chromatin associated proteins. BMC Genomics 2006, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Todd, M.A.; Picketts, D.J. Phf6 interacts with the nucleosome remodeling and deacetylation (NuRD) complex. J. Proteome Res. 2012, 11, 4326–4337. [Google Scholar] [CrossRef] [PubMed]
- Landais, S.; Quantin, R.; Rassart, E. Radiation leukemia virus common integration at the kis2 locus: Simultaneous overexpression of a novel noncoding rna and of the proximal phf6 gene. J. Virol 2005, 79, 11443–11456. [Google Scholar] [CrossRef] [PubMed]
- Vallee, D.; Chevrier, E.; Graham, G.E.; Lazzaro, M.A.; Lavigne, P.A.; Hunter, A.G.; Picketts, D.J. A novel phf6 mutation results in enhanced exon skipping and mild borjeson-forssman-lehmann syndrome. J. Med. Genet. 2004, 41, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Y.; Boisvert, F.M.; Gregor, P.; Cobley, A.; Lamond, A.I. Nopdb: Nucleolar proteome database--2008 update. Nucleic Acids Res. 2009, 37, D181–184. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.M.; Ahmad, Y.; Gierlinski, M.; Charriere, F.; Lamont, D.; Scott, M.; Barton, G.; Lamond, A.I. A quantitative spatial proteomics analysis of proteome turnover in human cells. MCP 2012, 11, M111 011429. [Google Scholar] [CrossRef] [PubMed]
- Voss, A.K.; Gamble, R.; Collin, C.; Shoubridge, C.; Corbett, M.; Gecz, J.; Thomas, T. Protein and gene expression analysis of phf6, the gene mutated in the borjeson-forssman-lehmann syndrome of intellectual disability and obesity. Gene Expr. Patterns 2007, 7, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Franzoni, E.; Booker, S.A.; Parthasarathy, S.; Rehfeld, F.; Grosser, S.; Srivatsa, S.; Fuchs, H.R.; Tarabykin, V.; Vida, I.; Wulczyn, F.G. Mir-128 regulates neuronal migration, outgrowth and intrinsic excitability via the intellectual disability gene phf6. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at ucsc. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O'Neil, J.; Neuberg, D.; Weng, A.P.; et al. Notch1 directly regulates c-myc and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Kraszewska, M.D.; Dawidowska, M.; Larmonie, N.S.; Kosmalska, M.; Sedek, L.; Szczepaniak, M.; Grzeszczak, W.; Langerak, A.W.; Szczepanski, T.; Witt, M. DNA methylation pattern is altered in childhood t-cell acute lymphoblastic leukemia patients as compared with normal thymic subsets: Insights into cpg island methylator phenotype in t-all. Leukemia 2012, 26, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; Van Der Meulen, J.; Wolfe, A.L.; Liu, X.; Mets, E.; Taghon, T.; Khan, A.A.; Setty, M.; Rondou, P.; Vandenberghe, P.; et al. A cooperative microrna-tumor suppressor gene network in acute t-cell lymphoblastic leukemia (t-all). Nat. Genet. 2011, 43, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Mets, E.; Van Peer, G.; Van der Meulen, J.; Boice, M.; Taghon, T.; Goossens, S.; Mestdagh, P.; Benoit, Y.; De Moerloose, B.; Van Roy, N.; et al. Microrna-128–3p is a novel oncomir targeting phf6 in t-cell acute lymphoblastic leukemia. Haematologica 2014. [Google Scholar]
- Borjeson, M.; Forssman, H.; Lehmann, O. An x-linked, recessively inherited syndrome characterized by grave mental deficiency, epilepsy, and endocrine disorder. Acta medica Scandinavica 1962, 171, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.; Lower, K.M.; White, S.M.; Delatycki, M.; Lampe, A.K.; Wright, M.; Smith, J.C.; Kerr, B.; Schelley, S.; Hoyme, H.E.; et al. The clinical picture of the borjeson-forssman-lehmann syndrome in males and heterozygous females with phf6 mutations. Clin. Genet. 2004, 65, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Gecz, J.; Turner, G.; Nelson, J.; Partington, M. The borjeson-forssman-lehman syndrome (bfls, mim #301900). Eur. J. Hum. Genet. 2006, 14, 1233–1237. [Google Scholar] [PubMed]
- Carter, M.T.; Picketts, D.J.; Hunter, A.G.; Graham, G.E. Further clinical delineation of the borjeson-forssman-lehmann syndrome in patients with phf6 mutations. Am. J. Med. Genet. A 2009, 149A, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Zweier, C.; Rittinger, O.; Bader, I.; Berland, S.; Cole, T.; Degenhardt, F.; Di Donato, N.; Graul-Neumann, L.; Hoyer, J.; Lynch, S.A.; et al. Females with de novo aberrations in phf6: Clinical overlap of borjeson-forssman-lehmann with coffin-siris syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166C, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.M.; Todd, M.A.; Kontny, U.; Neas, K.; Sullivan, M.J.; Hunter, A.G.; Picketts, D.J.; Kratz, C.P. T-cell acute lymphoblastic leukemia in association with borjeson-forssman-lehmann syndrome due to a mutation in phf6. Pediatr. Blood Cancer 2010, 55, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.; Lower, K.M.; Hennekam, R.C.; Van Esch, H.; Megarbane, A.; Lynch, S.A.; Turner, G.; Gecz, J. Mutation screening in borjeson-forssman-lehmann syndrome: Identification of a novel de novo phf6 mutation in a female patient. J. Med. Genet. 2006, 43, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Gedeon, A.K.; Kozman, H.M.; Robinson, H.; Pilia, G.; Schlessinger, D.; Turner, G.; Mulley, J.C. Refinement of the background genetic map of xq26-q27 and gene localisation for borjeson-forssman-lehmann syndrome. Am. J. Med. Genet. 1996, 64, 63–68. [Google Scholar] [CrossRef]
- Mathews, K.D.; Ardinger, H.H.; Nishimura, D.Y.; Buetow, K.H.; Murray, J.C.; Bartley, J.A. Linkage localization of borjeson-forssman-lehmann syndrome. Am. J. Med. Genet. 1989, 34, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.; Gedeon, A.; Mulley, J.; Sutherland, G.; Rae, J.; Power, K.; Arthur, I. Borjeson-forssman-lehmann syndrome: Clinical manifestations and gene localization to xq26–27. Am. J. Med. Genet. 1989, 34, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.E.; Schwartz, C.; Schroer, R.J. X-Linked Mental Retardation (Oxford Monographs on Medical Genetics, No. 39); Oxford University Press: New York, USA, 2000; pp. 134–138. [Google Scholar]
- Kosho, T.; Miyake, N.; Carey, J.C. Coffin-siris syndrome and related disorders involving components of the baf (mswi/snf) complex: Historical review and recent advances using next generation sequencing. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166C, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Okamoto, N.; Coffin-Siris Syndrome International. Genotype-phenotype correlation of coffin-siris syndrome caused by mutations in smarcb1, smarca4, smarce1, and arid1a. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166C, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Coffin, G.S.; Siris, E. Mental retardation with absent fifth fingernail and terminal phalanx. Am. J. Dis. Children 1970, 119, 433–439. [Google Scholar] [CrossRef]
- Tsurusaki, Y.; Okamoto, N.; Ohashi, H.; Kosho, T.; Imai, Y.; Hibi-Ko, Y.; Kaname, T.; Naritomi, K.; Kawame, H.; Wakui, K.; et al. Mutations affecting components of the swi/snf complex cause coffin-siris syndrome. Nat. Genet. 2012, 44, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, D.; Bogershausen, N.; Beleggia, F.; Steiner-Haldenstatt, S.; Pohl, E.; Li, Y.; Milz, E.; Martin, M.; Thiele, H.; Altmuller, J.; et al. A comprehensive molecular study on coffin-siris and nicolaides-baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet. 2013, 22, 5121–5135. [Google Scholar] [CrossRef] [PubMed]
- Zweier, C.; Kraus, C.; Brueton, L.; Cole, T.; Degenhardt, F.; Engels, H.; Gillessen-Kaesbach, G.; Graul-Neumann, L.; Horn, D.; Hoyer, J.; et al. A new face of borjeson-forssman-lehmann syndrome? De novo mutations in phf6 in seven females with a distinct phenotype. J. Med. Genet. 2013, 50, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Berland, S.; Alme, K.; Brendehaug, A.; Houge, G.; Hovland, R. Phf6 deletions may cause borjeson-forssman-lehmann syndrome in females. Mol. Syndromol 2011, 1, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, N.; Isidor, B.; Lopez Cazaux, S.; Le Caignec, C.; Klink, B.; Kraus, C.; Schrock, E.; Hackmann, K. Distinct phenotype of phf6 deletions in females. Eur J. Med. Genet. 2014, 57, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, M.; Chevrier, E.; Mustonen, A.; Picketts, D.J. Borjeson-forssman-lehmann syndrome due to a novel plant homeodomain zinc finger mutation in the phf6 gene. J. Child. Neurol 2009, 24, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Baumstark, A.; Lower, K.M.; Sinkus, A.; Andriuskeviciute, I.; Jurkeniene, L.; Gecz, J.; Just, W. Novel phf6 mutation p.D333del causes borjeson-forssman-lehmann syndrome. J. Med. Genet. 2003, 40, e50. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Haferlach, C.; Weissmann, S.; Roller, A.; Schindela, S.; Poetzinger, F.; Stadler, K.; Bellos, F.; Kern, W.; Haferlach, T.; et al. The molecular profile of adult t-cell acute lymphoblastic leukemia: Mutations in runx1 and dnmt3a are associated with poor prognosis in t-all. Genes Chromosomes Cancer 2013, 52, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qiu, H.; Jiang, H.; Wu, L.; Dong, S.; Pan, J.; Wang, W.; Ping, N.; Xia, J.; Sun, A.; et al. Mutations of phf6 are associated with mutations of notch1, jak1 and rearrangement of set-nup214 in t-cell acute lymphoblastic leukemia. Haematologica 2011, 96, 1808–1814. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.J.; Kim, Y.R.; Lee, S.H. Somatic mutation of phf6 gene in t-cell acute lymphoblatic leukemia, acute myelogenous leukemia and hepatocellular carcinoma. Acta Oncol. 2012, 51, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Huh, H.J.; Lee, S.H.; Yoo, K.H.; Sung, K.W.; Koo, H.H.; Jang, J.H.; Kim, K.; Kim, S.J.; Kim, W.S.; Jung, C.W.; et al. Gene mutation profiles and prognostic implications in korean patients with t-lymphoblastic leukemia. Ann. Hematol 2013, 92, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Tanaka, H.; Naoe, Y.; Taniuchi, I. Transcriptional control of t-cell development. Int. Immunol. 2011, 23, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Ferrando, A. The molecular basis of t cell acute lymphoblastic leukemia. J. Clin. Invest. 2012, 122, 3398–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in t cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef]
- De Keersmaecker, K.; Real, P.J.; Gatta, G.D.; Palomero, T.; Sulis, M.L.; Tosello, V.; Van Vlierberghe, P.; Barnes, K.; Castillo, M.; Sole, X.; et al. The tlx1 oncogene drives aneuploidy in t cell transformation. Nat. Med. 2010, 16, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.t.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of notch1 in human t cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.P.; Gonen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, H.; Chen, Z.; Wang, Q.; Zhao, Y.; Chen, S. Somatic mutations of phf6 in patients with chronic myeloid leukemia in blast crisis. Leuk Lymphoma 2013, 54, 671–672. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Lawton, L.N.; Soto-Feliciano, Y.M.; Pritchard, J.R.; Joughin, B.A.; Ehrenberger, T.; Fenouille, N.; Zuber, J.; Williams, R.T.; Young, R.A.; et al. A genome-scale in vivo loss-of-function screen identifies phf6 as a lineage-specific regulator of leukemia cell growth. Genes Dev. 2015, 29, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Leung, J.W.; Gong, Z.; Feng, L.; Shi, X.; Chen, J. Phf6 regulates cell cycle progression by suppressing ribosomal rna synthesis. J. Biol. Chem. 2013, 288, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, Y.; Watanabe, H.; Inoue, J.; Takeda, N.; Sakata, J.; Mishima, Y.; Hitomi, J.; Yamamoto, T.; Utsuyama, M.; Niwa, O.; et al. Bcl11b is required for differentiation and survival of alphabeta t lymphocytes. Nat. Immunol. 2003, 4, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Aasland, R.; Gibson, T.J.; Stewart, A.F. The phd finger: Implications for chromatin-mediated transcriptional regulation. Trends Biochem. Sci 1995, 20, 56–59. [Google Scholar] [CrossRef]
- Capili, A.D.; Schultz, D.C.; Rauscher, I.F.; Borden, K.L. Solution structure of the phd domain from the kap-1 corepressor: Structural determinants for phd, ring and lim zinc-binding domains. Embo J. 2001, 20, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ilin, S.; Wang, W.; Duncan, E.M.; Wysocka, J.; Allis, C.D.; Patel, D.J. Molecular basis for site-specific read-out of histone h3k4me3 by the bptf phd finger of nurf. Nature 2006, 442, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Pena, P.; Lan, F.; Kaadige, M.R.; et al. Ing2 phd domain links histone h3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [PubMed]
- Mansfield, R.E.; Musselman, C.A.; Kwan, A.H.; Oliver, S.S.; Garske, A.L.; Davrazou, F.; Denu, J.M.; Kutateladze, T.G.; Mackay, J.P. Plant homeodomain (phd) fingers of chd4 are histone h3-binding modules with preference for unmodified h3k4 and methylated h3k9. J. Biol. Chem. 2011, 286, 11779–11791. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Kaynak, B.; Forster, U.B.; Tonjes, M.; Fischer, J.J.; Grimm, C.; Schlesinger, J.; Just, S.; Dunkel, I.; Krueger, T.; et al. Regulation of muscle development by dpf3, a novel histone acetylation and methylation reader of the baf chromatin remodeling complex. Genes Dev. 2008, 22, 2370–2384. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, Q.; Li, S.; Plotnikov, A.N.; Walsh, M.J.; Zhou, M.M. Mechanism and regulation of acetylated histone binding by the tandem phd finger of dpf3b. Nature 2010, 466, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qin, S.; Zhang, J.; Ji, P.; Shi, Y.; Wu, J. Solution structure of an atypical phd finger in brpf2 and its interaction with DNA. J. Struct. Biol. 2012, 180, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, F.; Ruan, K.; Zhang, J.; Mei, Y.; Wu, J.; Shi, Y. Structural and functional insights into the human borjeson-forssman-lehmann syndrome-associated protein phf6. J. Biol. Chem. 2014, 289, 10069–10083. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into rna biology from an atlas of mammalian mrna-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Guy, J.; Ramsahoye, B.; Wilson, V.A.; Bird, A. Closely related proteins mbd2 and mbd3 play distinctive but interacting roles in mouse development. Genes Dev. 2001, 15, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Wade, P.A. Cancer biology and nurd: A multifaceted chromatin remodelling complex. Nat. Rev. Cancer 2011, 11, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Potts, R.C.; Zhang, P.; Wurster, A.L.; Precht, P.; Mughal, M.R.; Wood, W.H., 3rd; Zhang, Y.; Becker, K.G.; Mattson, M.P.; Pazin, M.J. Chd5, a brain-specific paralog of mi2 chromatin remodeling enzymes, regulates expression of neuronal genes. PLoS One 2011, 6, e24515. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Hazan, I.; Zhang, J.; Ng, S.Y.; Naito, T.; Snippert, H.J.; Heller, E.J.; Qi, X.; Lawton, L.N.; Williams, C.J.; et al. The role of the chromatin remodeler mi-2beta in hematopoietic stem cell self-renewal and multilineage differentiation. Genes Dev. 2008, 22, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.; Latos, P.; Hynes-Allen, A.; Loos, R.; Leaford, D.; O'Shaughnessy, A.; Mosaku, O.; Signolet, J.; Brennecke, P.; Kalkan, T.; et al. Nurd suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell. Stem Cell 2012, 10, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Yang, Y.; Hemberg, M.; Yoshida, T.; Cho, H.Y.; Murphy, J.P.; Fioravante, D.; Regehr, W.G.; Gygi, S.P.; Georgopoulos, K.; et al. Promoter decommissioning by the nurd chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron 2014, 83, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.; Salmon-Divon, M.; Dvinge, H.; Hynes-Allen, A.; Balasooriya, G.; Leaford, D.; Behrens, A.; Bertone, P.; Hendrich, B. Nurd-mediated deacetylation of h3k27 facilitates recruitment of polycomb repressive complex 2 to direct gene repression. EMBO J. 2012, 31, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jackson, A.F.; Naito, T.; Dose, M.; Seavitt, J.; Liu, F.; Heller, E.J.; Kashiwagi, M.; Yoshida, T.; Gounari, F.; et al. Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat. Immunol. 2012, 13, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.K.; Hassig, C.A.; Schnitzler, G.R.; Kingston, R.E.; Schreiber, S.L. Chromatin deacetylation by an atp-dependent nucleosome remodelling complex. Nature 1998, 395, 917–921. [Google Scholar] [PubMed]
- Zhang, Y.; LeRoy, G.; Seelig, H.P.; Lane, W.S.; Reinberg, D. The dermatomyositis-specific autoantigen mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 1998, 95, 279–289. [Google Scholar] [CrossRef]
- Williams, C.J.; Naito, T.; Arco, P.G.; Seavitt, J.R.; Cashman, S.M.; De Souza, B.; Qi, X.; Keables, P.; Von Andrian, U.H.; Georgopoulos, K. The chromatin remodeler mi-2beta is required for cd4 expression and t cell development. Immunity 2004, 20, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Chen, Y.; Sun, Y.; Yang, F.; Yu, W.; Liang, J.; Sun, L.; Yang, X.; Shi, L.; et al. Lsd1 is a subunit of the nurd complex and targets the metastasis programs in breast cancer. Cell 2009, 138, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Molnar, A.; Georgopoulos, K. The ikaros gene encodes a family of functionally diverse zinc finger DNA-binding proteins. Mol. Cell. Biol. 1994, 14, 8292–8303. [Google Scholar] [PubMed]
- Yamashita, K.; Sato, A.; Asashima, M.; Wang, P.C.; Nishinakamura, R. Mouse homolog of sall1, a causative gene for townes-brocks syndrome, binds to a/t-rich sequences in pericentric heterochromatin via its c-terminal zinc finger domains. Genet. Cells 2007, 12, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Lauberth, S.M.; Bilyeu, A.C.; Firulli, B.A.; Kroll, K.L.; Rauchman, M. A phosphomimetic mutation in the sall1 repression motif disrupts recruitment of the nucleosome remodeling and deacetylase complex and repression of gbx2. J. Biol. Chem. 2007, 282, 34858–34868. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Di Lena, P.; Schaffer, L.; Head, S.R.; Baldi, P.; Thomas, E.A. Genome-wide identification of bcl11b gene targets reveals role in brain-derived neurotrophic factor signaling. PLoS One 2011, 6, e23691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Mejia, L.A.; Huang, J.; Valnegri, P.; Bennett, E.J.; Anckar, J.; Jahani-Asl, A.; Gallardo, G.; Ikeuchi, Y.; Yamada, T.; et al. The x-linked intellectual disability protein phf6 associates with the paf1 complex and regulates neuronal migration in the mammalian brain. Neuron 2013, 78, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Cayrou, C.; Ullah, M.; Landry, A.J.; Cote, V.; Selleck, W.; Lane, W.S.; Tan, S.; Yang, X.J.; Cote, J. Ing tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell. 2006, 21, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.S.; Helton, E.S.; Banerjee, S.; Venable, M.; Johnson, L.; Schoeb, T.R.; Kesterson, R.A.; Crawford, D.F. G2e3 is a dual function ubiquitin ligase required for early embryonic development. J. Biol. Chem. 2008, 283, 22304–22315. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of histone lysine trimethylation by the jmjd2 family of histone demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Mori, T.; Tada, S.; Krajewski, W.; Rozovskaia, T.; Wassell, R.; Dubois, G.; Mazo, A.; Croce, C.M.; Canaani, E. All-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol. Cell. 2002, 10, 1119–1128. [Google Scholar] [CrossRef]
- Mo, R.; Rao, S.M.; Zhu, Y.J. Identification of the mll2 complex as a coactivator for estrogen receptor alpha. J. Biol. Chem. 2006, 281, 15714–15720. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. Ptip associates with mll3- and mll4-containing histone h3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Baxter, E.M.; Van Doren, M. Phf7 controls male sex determination in the drosophila germline. Dev. Cell 2012, 22, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Clarke, E.; Rahman, N.; Page, N.; Rolph, M.S.; Stewart, G.J.; Jones, G.J. Functional characterization of the atopy-associated gene phf11. J. Allergy Clin. Immunol. 2008, 121, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Takebe, A.; Ono, Y.; Imai, T.; Nakao, K.; Nishikawa, S.; Era, T. Phf14, a novel regulator of mesenchyme growth via platelet-derived growth factor (pdgf) receptor-alpha. J. Biol. Chem. 2012, 287, 27983–27996. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Saifi, G.M.; Shaw, C.J.; Walz, K.; Fonseca, P.; Wilson, M.; Potocki, L.; Lupski, J.R. Mutations of rai1, a phd-containing protein, in nondeletion patients with smith-magenis syndrome. Human Genet. 2004, 115, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Rekdal, C.; Sjottem, E.; Johansen, T. The nuclear factor spbp contains different functional domains and stimulates the activity of various transcriptional activators. J. Biol. Chem. 2000, 275, 40288–40300. [Google Scholar] [CrossRef] [PubMed]
- Lauberth, S.M.; Rauchman, M. A conserved 12-amino acid motif in sall1 recruits the nucleosome remodeling and deacetylase corepressor complex. J. Biol. Chem. 2006, 281, 23922–23931. [Google Scholar] [CrossRef] [PubMed]
- Lejon, S.; Thong, S.Y.; Murthy, A.; AlQarni, S.; Murzina, N.V.; Blobel, G.A.; Laue, E.D.; Mackay, J.P. Insights into association of the nurd complex with fog-1 from the crystal structure of an rbap48.Fog-1 complex. J. Biol. Chem. 2011, 286, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, F.; Zhang, B.; Li, S.; Wu, J.; Shi, Y. Structural basis of plant homeodomain finger 6 (phf6) recognition by the retinoblastoma binding protein 4 (rbbp4) component of the nucleosome remodeling and deacetylase (nurd) complex. J. Biol. Chem. 2015, 290, 6630–6638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Smith, A.D.t.; Renfrow, M.B.; Schneider, D.A. The rna polymerase-associated factor 1 complex (paf1c) directly increases the elongation rate of rna polymerase i and is required for efficient regulation of rrna synthesis. J. Biol. Chem. 2010, 285, 14152–14159. [Google Scholar] [CrossRef] [PubMed]
- Learned, R.M.; Learned, T.K.; Haltiner, M.M.; Tjian, R.T. Human rrna transcription is modulated by the coordinate binding of two factors to an upstream control element. Cell 1986, 45, 847–857. [Google Scholar] [CrossRef]
- Bell, S.P.; Learned, R.M.; Jantzen, H.M.; Tjian, R. Functional cooperativity between transcription factors ubf1 and sl1 mediates human ribosomal rna synthesis. Science 1988, 241, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. Atm and atr substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl Acad Sci U S A 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed]
- Mayya, V.; Lundgren, D.H.; Hwang, S.I.; Rezaul, K.; Wu, L.; Eng, J.K.; Rodionov, V.; Han, D.K. Quantitative phosphoproteomic analysis of t cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal. 2009, 2, ra46. [Google Scholar] [CrossRef] [PubMed]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative phosphoproteomics identifies substrates and functional modules of aurora and polo-like kinase activities in mitotic cells. Sci. Signal. 2011, 4, rs5. [Google Scholar] [CrossRef] [PubMed]
- Kruhlak, M.; Crouch, E.E.; Orlov, M.; Montano, C.; Gorski, S.A.; Nussenzweig, A.; Misteli, T.; Phair, R.D.; Casellas, R. The atm repair pathway inhibits rna polymerase i transcription in response to chromosome breaks. Nature 2007, 447, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Identification of a consensus motif for plk (polo-like kinase) phosphorylation reveals myt1 as a plk1 substrate. J. Biol. Chem. 2003, 278, 25277–25280. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.; Cantley, L.C.; Yaffe, M.B. Proteomic screen finds pser/pthr-binding domain localizing plk1 to mitotic substrates. Science 2003, 299, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Grosstessner-Hain, K.; Hegemann, B.; Novatchkova, M.; Rameseder, J.; Joughin, B.A.; Hudecz, O.; Roitinger, E.; Pichler, P.; Kraut, N.; Yaffe, M.B.; et al. Quantitative phospho-proteomics to investigate the polo-like kinase 1-dependent phospho-proteome. MCP 2011, 10, M111 008540. [Google Scholar] [PubMed]
- Chi, Y.; Welcker, M.; Hizli, A.A.; Posakony, J.J.; Aebersold, R.; Clurman, B.E. Identification of cdk2 substrates in human cell lysates. Genome Biol. 2008, 9, R149. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Hegarat, N.; Wills, K.L.; Furlan, C.; Bukowski-Wills, J.C.; Hochegger, H.; Rappsilber, J. Proteomics of a fuzzy organelle: Interphase chromatin. EMBO J. 2014, 33, 648–664. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nature Rev. Drug Discovery 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, L.; Trere, D.; Derenzini, M. Nucleolus, ribosomes, and cancer. Am. J. Pathol. 2008, 173, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sif, S.; Jones, B.; Jackson, A.; Koipally, J.; Heller, E.; Winandy, S.; Viel, A.; Sawyer, A.; Ikeda, T.; et al. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity 1999, 10, 345–355. [Google Scholar] [CrossRef]
- Bottardi, S.; Mavoungou, L.; Bourgoin, V.; Mashtalir, N.; Affar el, B.; Milot, E. Direct protein interactions are responsible for ikaros-gata and ikaros-cdk9 cooperativeness in hematopoietic cells. Mol. Cell. Biol. 2013, 33, 3064–3076. [Google Scholar] [CrossRef] [PubMed]
- Bottardi, S.; Mavoungou, L.; Pak, H.; Daou, S.; Bourgoin, V.; Lakehal, Y.A.; Affar el, B.; Milot, E. The ikaros interaction with a complex including chromatin remodeling and transcription elongation activities is required for hematopoiesis. PLoS Genet. 2014, 10, e1004827. [Google Scholar] [CrossRef] [PubMed]
- Shimbo, T.; Du, Y.; Grimm, S.A.; Dhasarathy, A.; Mav, D.; Shah, R.R.; Shi, H.; Wade, P.A. Mbd3 localizes at promoters, gene bodies and enhancers of active genes. PLoS Genet. 2013, 9, e1004028. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Ling, T.; Zhou, Y.; Feng, W.; Zhu, Q.; Stunnenberg, H.G.; Grummt, I.; Tao, W. The chromatin remodeling complex nurd establishes the poised state of rrna genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl. Acad. Sci. USA 2012, 109, 8161–8166. [Google Scholar] [CrossRef] [PubMed]
- Ling, T.; Xie, W.; Luo, M.; Shen, M.; Zhu, Q.; Zong, L.; Zhou, T.; Gu, J.; Lu, Z.; Zhang, F.; et al. Chd4/nurd maintains demethylation state of rdna promoters through inhibiting the expression of the rdna methyltransferase recruiter tip5. Biochem. Biophys. Res. Commun. 2013, 437, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Wittner, M.; Babl, V.; Perez-Fernandez, J.; Tschochner, H.; Griesenbeck, J. Chromatin states at ribosomal DNA loci. Biochimica et biophysica acta 2013, 1829, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Montanaro, L.; Trere, D. What the nucleolus says to a tumour pathologist. Histopathology 2009, 54, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Girardi, T.; De Keersmaecker, K. T-all: All a matter of translation? Haematologica 2015, 100, 293–295. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, K.; Sulima, S.O.; Dinman, J.D. Ribosomopathies and the paradox of cellular hypo- to hyperproliferation. Blood 2015, 125, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.R.; McIntosh, K.B. How common are extraribosomal functions of ribosomal proteins? Mol. Cell. 2009, 34, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Smith, S.C.; Youssef, M.N.; Zheng, J.J.; Hagg, T.; Hetman, M. Rna polymerase 1-driven transcription as a mediator of bdnf-induced neurite outgrowth. J. Biol. Chem. 2011, 286, 4357–4363. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Sanij, E.; Rothblum, L.I.; Hannan, R.D.; Pearson, R.B. Dysregulation of rna polymerase i transcription during disease. Biochimica et biophysica acta 2013, 1829, 342–360. [Google Scholar] [CrossRef] [PubMed]
- Hetman, M.; Pietrzak, M. Emerging roles of the neuronal nucleolus. Trends Neurosci. 2012, 35, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in x-linked mecp2, encoding methyl-cpg-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Santoro, R.; Grummt, I. Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription. Mol. Cell. 2001, 8, 719–725. [Google Scholar] [CrossRef]
- Brero, A.; Easwaran, H.P.; Nowak, D.; Grunewald, I.; Cremer, T.; Leonhardt, H.; Cardoso, M.C. Methyl cpg-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 2005, 169, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.K.; Gonzales, M.L.; Leung, K.N.; Yasui, D.H.; Schroeder, D.I.; Dunaway, K.; LaSalle, J.M. Mecp2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol. Dis. 2011, 43, 190–200. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todd, M.A.M.; Ivanochko, D.; Picketts, D.J. PHF6 Degrees of Separation: The Multifaceted Roles of a Chromatin Adaptor Protein. Genes 2015, 6, 325-352. https://doi.org/10.3390/genes6020325
Todd MAM, Ivanochko D, Picketts DJ. PHF6 Degrees of Separation: The Multifaceted Roles of a Chromatin Adaptor Protein. Genes. 2015; 6(2):325-352. https://doi.org/10.3390/genes6020325
Chicago/Turabian StyleTodd, Matthew A.M., Danton Ivanochko, and David J. Picketts. 2015. "PHF6 Degrees of Separation: The Multifaceted Roles of a Chromatin Adaptor Protein" Genes 6, no. 2: 325-352. https://doi.org/10.3390/genes6020325
APA StyleTodd, M. A. M., Ivanochko, D., & Picketts, D. J. (2015). PHF6 Degrees of Separation: The Multifaceted Roles of a Chromatin Adaptor Protein. Genes, 6(2), 325-352. https://doi.org/10.3390/genes6020325