
Alternative Okazaki Fragment Ligation Pathway by DNA Ligase III


Abstract
:1. Introduction

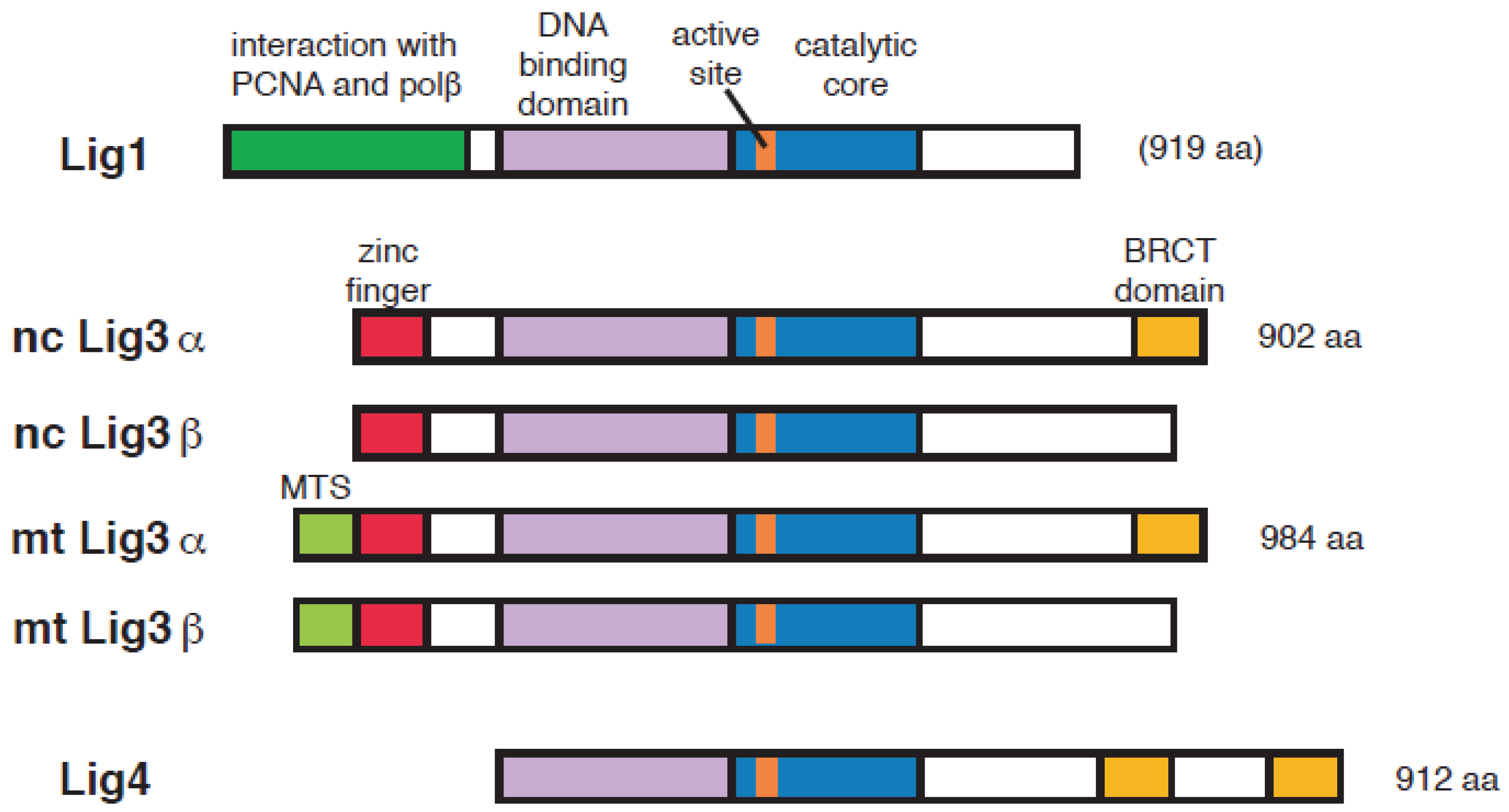
2. DNA Ligase Responsible for the Ligation of Okazaki Fragments
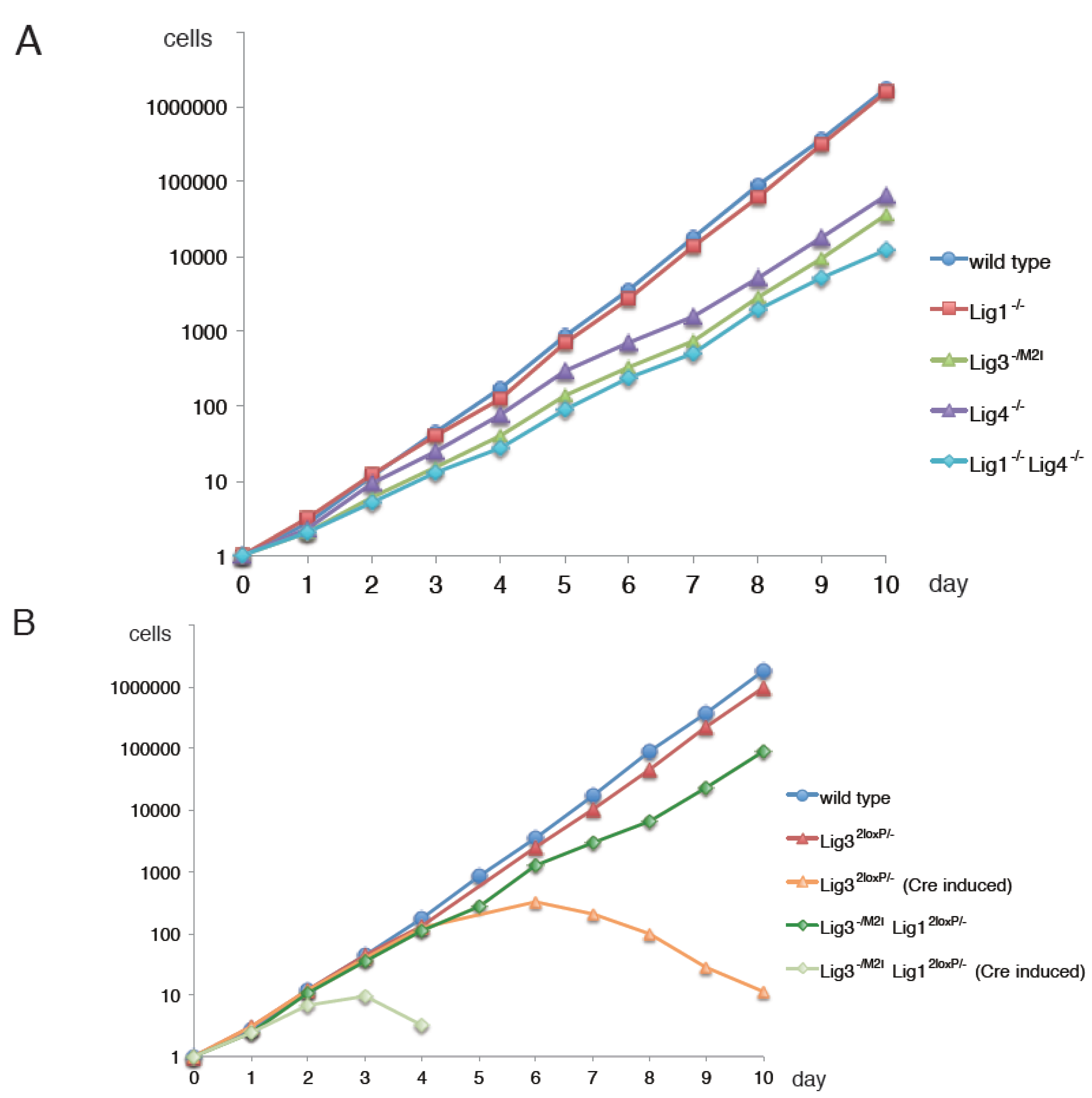
3. Has Lig3 A Function in DNA Replication Outside Mitochondria?
4. Why is Mitochondrial Lig3 Essential?

5. Alternative Okazaki-Fragment Ligation Pathway by Lig3
6. Lig3 as a Universal DNA Ligase
7. Conclusions

{kind=link}
{kind=link}
| Concept | Lig1 | Lig3 | Lig4 |
|---|---|---|---|
| classical concept | DNA replication | BER (short patch) | NHEJ |
| BER (long patch) | mitochondria | V(D)J recombination | |
| HR | class switch recombination | ||
| NER | |||
| new concept | DNA replication | DNA replication | NHEJ |
| BER (long patch) | BER (short patch) | V(D)J recombination | |
| HR | HR | class switch recombination | |
| NER | NER | ||
| B-NHEJ | B-NHEJ | ||
| (V(D)J recombination?) | (V(D)J recombination?) | ||
| (class switch recombination?) | (class switch recombination?) | ||
| mitochondria |
8. Future Perspectives
- As Lig1 knockout DT40 cells proliferate normally, it must be assumed that Lig3 is recruited to DNA replication sites as effectively as Lig1. How is this achieved and how do cells chose between the two DNA ligases?
- It has been reported that Lig1 and Lig3 have a significant contribution to translocation formation in rodent cell systems [59,60,61]. In human cells, classical NHEJ, and thus Lig4, has a bulk contribution to chromosome translocation formation initiated by DSB induced by designer nucleases [62]. However, chromosome translocations generated in human cells exposed during the G2-phase to IR, which induces DSB randomly distributed throughout the genome, largely rely on Lig1 and Lig3 as is the case in rodent cells [63]. This raises the question of the evolutionary significance of Lig3 in chromosomal translocation formation, and thus in the development of cancer.
- Why mitochondria are served in vertebrates preferentially by Lig3 over Lig1, and why did Lig1 lose this duty during evolution?
- As Xrcc1 is not necessary for the mitochondrial functions of Lig3, it can be inferred that Lig3 can also function without this cofactor. This raises the question as to whether certain aspects of the nuclear functions of Lig3 are also Xrcc1-independent.
- Lig3 may have important partners in addition to Xrcc1. Indeed, Lig3 may change partners depending upon the process it is involved, and possibly also the cellular physiology. What are these partners and what is the functional significance of the corresponding interactions?
- Although Lig2 is considered at present a biochemical artefact, the possibility should be left open that it is more than a random degradation product of Lig3: it may well be the fifth variant of Lig3 endowed with specific but as of yet uncharacterized functions.
- Is Lig1 knockout in the mouse lethal if mediated by Cre/lox P in adult stage, where Lig3 should be able to fully compensate?
Acknowledgments
Conflicts of Interest
Abbreviations
| BER | Base excision repair |
| BRCA1 | breast cancer susceptibility gene 1 |
| BRCT | BRCA1 C terminus |
| B-NHEJ | back up-NHEJ |
| C-NHEJ | classical non-homologous end-joining |
| Lig1 | DNA ligase 1 |
| Lig2 | DNA ligase 2 |
| Lig3 | DNA ligase 3 |
| Lig4 | DNA ligase 4 |
| IR | ionizing radiation |
| DSB | double strand break |
| HRR | homologous recombination repair |
| MEF | mouse embryonic fibroblasts |
| MTS | mitochondria target sequence |
| NER | nucleotide excision repair |
| SSBR | single strand break repair |
| PCNA | proliferating cell nuclear antigen |
| XRCC1 | X-ray repair cross-complementing protein 1 |
References
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. Biochem. 2008, 77, 313–338. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, A.E.; Mackey, Z.B. Structure and function of mammalian DNA ligases. Mutat. Res. 1998, 407, 1–9. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D.E. Mammalian DNA ligases. Annu. Rev. Biochem. 1992, 61, 251–281. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Bednar, T.; Wang, M.; Paul, K.; Mladenov, E.; Bencsik-Theilen, A.A.; Iliakis, G. Functional redundancy between DNA ligases I and III in DNA replication in vertebrate cells. Nucleic Acids Res. 2012, 40, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Husain, I.; Tomkinson, A.E.; Burkhart, W.A.; Moyer, M.B.; Ramos, W.; Mackey, Z.B.; Besterman, J.M.; Chen, J. Purification and characterization of DNA ligase III from bovine testes. Homology with DNA ligase II and vaccinia DNA ligase. J. Biol. Chem. 1995, 270, 9683–9690. [Google Scholar] [PubMed]
- Petrini, J.H.J.; Xiao, Y.; Weaver, D.T. DNA ligase I mediates essential functions in mammalian cells. Mol. Cell. Biol. 1995, 15, 4303–4308. [Google Scholar] [PubMed]
- Bentley, D.J.; Selfridge, J.; Millar, J.K.; Samuel, K.; Hole, N.; Ansell, J.D.; Melton, D.W. DNA ligase I is required for fetal liver erythropoiesis but is not essential for mammlian cell viability. Nat. Genet. 1996, 13, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Puebla-Osorio, N.; Lacey, D.B.; Alt, F.W.; Zhu, C. Early embryonic lethality due to targeted inactivation of DNA ligase III. Mol. Cell. Biol. 2006, 26, 3935–3941. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Stamp, G.; Rosewell, I.; Denzel, A.; Lindahl, T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol. 1998, 8, 1395–1398. [Google Scholar] [CrossRef]
- Frank, K.M.; Sekiguchi, J.M.; Seidl, K.J.; Swat, W.; Rathbun, G.A.; Cheng, H.-L.; Davidson, L.; Kangaloo, L.; Alt, F.W. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 1998, 396, 173–177. [Google Scholar] [PubMed]
- Wilson, T.E.; Grawunder, U.; Lieber, M.R. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature 1997, 388, 495–498. [Google Scholar] [PubMed]
- Montecucco, A.; Rossi, R.; Levin, D.S.; Gary, R.; Park, M.S.; Motycka, T.A.; Ciarrocchi, G.; Villa, A.; Biamonti, G.; Tomkinson, A.E. DNA ligase I is recruited to sites of DNA replication by an interaction with proliferating cell nuclear antigen: Identification of a common targeting mechanism for the assembly of replication factories. EMBO J. 1998, 17, 3786–3795. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Savini, E.; Weighardt, F.; Rossi, R.; Ciarrocchi, G.; Villa, A.; Biamonti, G. The N-terminal domain of human DNA ligase I contains the nuclear localization signal and directs the enzyme to sites of DNA replication. EMBO J. 1995, 14, 5379–5386. [Google Scholar] [PubMed]
- Johnston, L.H. The DNA repair capability of CDC9, the Saccharomyces cerevisiae mutant defective in DNA ligase. Mol. Gen. Genet. 1979, 170, 89–92. [Google Scholar] [PubMed]
- Johnston, L.H.; Nasmyth, K.A. Saccharomyces cerevisiae cell cycle mutant CDC9 is defective in DNA ligase. Nature 1978, 274, 891–893. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H. Saccharomyces cerevisiae cell cycle. Bacteriol. Rev. 1974, 38, 164–198. [Google Scholar] [PubMed]
- Bentley, D.J.; Harrison, C.; Ketchen, A-M.; Redhead, N.J.; Samuel, K.; Waterfall, M.; Ansell, J.D.; Melton, D.W. DNA ligase I null mouse cells show normal DNA repair activity but altered DNA replication and reduced genome stability. J. Cell Sci. 2002, 115, 1551–1561. [Google Scholar] [PubMed]
- Barnes, D.E.; Tomkinson, A.E.; Lehmann, A.R.; Webster, A.D.B.; Lindahl, T. Mutations in the DNA ligase 1 gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell 1992, 69, 495–503. [Google Scholar] [CrossRef]
- Webster, A.D.B.; Barnes, D.E.; Arlett, C.F.; Lehmann, A.R.; Lindahl, T. Growth retardation and immunodeficiency in a patient with mutations in the DNA ligase 1 gene. Lancet 1992, 339, 1508–1509. [Google Scholar] [CrossRef]
- Henderson, L.M.; Arlett, C.F.; Harcourt, S.A.; Lehmann, A.R.; Broughton, B.C. Cells from an immunodeficient patient (46BR) with a defect in DNA ligation are hypomutable but hypersensitive to the induction of sister chromatid exchanges. Proc. Natl. Acad. Sci. USA 1985, 82, 2044–2048. [Google Scholar] [CrossRef] [PubMed]
- Lönn, U.; Lönn, S.; Nylen, U.; Winblad, G. Altered formation of DNA replication intermediates in human 46 BR fibroblast cells hypersensitive to 3-aminobenzamide. Carcinogenesis 1989, 10, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Aboussekhra, A.; Biggerstaff, M.; Shivji, M.K.; Vilpo, J.A.; Moncollin, V.; Podust, V.M.; Protic, M.; Huebscher, U.; Egly, J.-M.; Wood, R.D. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 1995, 80, 859–868. [Google Scholar] [CrossRef]
- Levin, D.S.; McKenna, A.E.; Motycka, T.A.; Matsumoto, Y.; Tomkinson, A.E. Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr. Biol. 2000, 10, 919–922. [Google Scholar] [CrossRef]
- Lakshmipathy, U.; Campbell, C. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell. Biol. 1999, 19, 3869–3876. [Google Scholar] [PubMed]
- Perez-Jannotti, R.M.; Klein, S.M.; Bogenhagen, D.F. Two forms of mitochondrial DNA ligase III are produced in Xenopus laevis oocytes. J. Biol. Chem. 2001, 276, 48978–48987. [Google Scholar] [CrossRef] [PubMed]
- Mackey, Z.B.; Ramos, W.; Levin, D.S.; Walter, C.A.; McCarrey, J.R.; Tomkinson, A.E. An alternative splicing event which occurs in mouse pachytene spermatocytes generates a form of DNA ligase III with distinct biochemical properties that may function in meiotic recombination. Mol. Cell. Biol. 1997, 17, 989–998. [Google Scholar] [PubMed]
- Simsek, D.; Jasin, M. DNA ligase III: A spotty presence in eukaryotes, but an essential function where tested. Cell Cycle 2011, 10, 3636–3644. [Google Scholar] [CrossRef] [PubMed]
- Buerstedde, J.-M.; Takeda, S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell 1991, 67, 179–188. [Google Scholar] [CrossRef]
- Arakawa, H.; Hauschild, J.; Buerstedde, J.-M. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science 2002, 295, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Lodygin, D.; Buerstedde, J.-M. Mutant lox P vectors for selectable marker recycle and conditional knockouts. BMC Biotechnol. 2001. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Harvey, A.; Zimbric, J.; Wang, Y.; Nguyen, T.; Jackson, P.J.; Hendrickson, E.A. DNA ligase III and DNA ligase IV carry out genetically distinct forms of end joining in human somatic cells. DNA Repair 2014, 21, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Katyal, S.; Lee, Y.; Zhao, J.; Rehg, J.E.; Russell, H.R.; McKinnon, P.J. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 2011, 471, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Simsek, D.; Furda, A.; Gao, Y.; Artus, J.; Brunet, E.; Hadjantonakis, A.-K.; van Houten, B.; Shuman, S.; McKinnon, P.J.; Jasin, M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 2011, 471, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell. Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef] [PubMed]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Masani, S.; Hsieh, C.L.; Yu, K. DNA ligase I is not essential for mammalian cell viability. Cell Rep. 2014, 7, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.; Thomale, J.; Arakawa, H.; Iliakis, G. DNA ligases I and III support nucleotide excision repair in DT40 cells with similar efficiency. Photochem. Photobiol. 2015, in press. [Google Scholar]
- Cotner-Gohara, E.; Kim, I.-K.; Tomkinson, A.E.; Ellenberger, T. Two DNA-binding and nick recognition modules in human DNA ligase III. J. Biol. Chem. 2008, 283, 10764–10772. [Google Scholar] [CrossRef] [PubMed]
- Pascal, J.M.; O’Brien, P.J.; Tomkinson, A.E.; Ellenberger, T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature 2004, 432, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.C.; Joseph, C.; Rahn, H.-P.; Reusch, R.; Nadal-Ginard, B.; Leonhardt, H. Mapping and use of a sequence that targets DNA ligase I to sites of DNA replication in vivo. J. Cell Biol. 1997, 139, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Le Chalony, C.; Hoffschir, F.; Gauthier, L.R.; Gross, J.; Biard, D.S.; Boussin, F.D.; Pennaneach, V. Partial complementation of a DNA ligase I deficiency by DNA ligase III and its impact on cell survival and telomere stability in mammalian cells. Cell Mol. Life Sci. 2012, 69, 2933–2949. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Otterlei, M.; Wong, H.-K.; Tomkinson, A.E.; Wilson, D.M., III. XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 2004, 32, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Windhofer, F.; Wu, W.; Iliakis, G. Low levels of DNA ligases III and IV sufficient for effective NHEJ. J. Cell. Physiol. 2007, 213, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, M.; Wu, W.; Singh, S.K.; Mussfeldt, T.; Iliakis, G. Repair of radiation induced DNA double strand breaks by backup NHEJ is enhanced in G2. DNA Repair 2008, 7, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Wang, H.; Perrault, A.R.; Boecker, W.; Rosidi, B.; Windhofer, F.; Wu, W.; Guan, J.; Terzoudi, G.; Pantelias, G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004, 104, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. 2011, 711, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.T.; Boboila, C.; Souza, E.K.; Franco, S.; Hickernell, T.R.; Murphy, M.; Gumaste, S.; Geyer, M.; Zarrin, A.A.; Manis, J.P.; et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 2007, 449, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Nussenzweig, A.; Nussenzweig, M.C. A backup DNA repair pathway moves to the forefront. Cell 2007, 131, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Corneo, B.; Wendland, R.L.; Deriano, L.; Cui, X.; Klein, I.A.; Wong, S.Y.; Arnal, S.; Holub, A.J.; Weller, G.R.; Pancake, B.A.; et al. Rag mutations reveal robust alternative end joining. Nature 2007, 449, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Shull, E.R.; Lee, Y.; Nakane, H.; Stracker, T.H.; Zhao, J.; Russell, H.R.; Petrini, J.H.; McKinnon, P.J. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes Dev. 2009, 23, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rosidi, B.; Perrault, R.; Wang, M.; Zhang, L.; Windhofer, F.; Iliakis, G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005, 65, 4020–4030. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.; Wang, M.; Mladenov, E.; Bencsik-Theilen, A.; Bednar, T.; Wu, W.; Arakawa, H.; Iliakis, G. DNA ligases I and III cooperate in alternative non-homologous end-joining in vertebrates. PLoS ONE 2013, 8, e59505. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.E.; Oh, S.; Grimstead, J.W.; Zimbric, J.; Roger, L.; Heppel, N.H.; Ashelford, K.E.; Liddiard, K.; Hendrickson, E.A.; Baird, D.M. Escape from telomere-driven crisis is DNA ligase III dependent. Cell Rep. 2014, 8, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.F.; Fousteri, M.I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase IIIα in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Abdou, I.; Poirier, G.G.; Hendzel, M.J.; Weinfeld, M. DNA ligase III acts as a DNA strand break sensor in the cellular orchestration of DNA strand break repair. Nucleic Acids Res. 2015, 43, 875–892. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Iliakis, G. Efficient homologous recombination repair in mutants relying exclusively on DNA ligase III for all their ligation requirements. Unpublished work. 2015. [Google Scholar]
- Simsek, D.; Brunet, E.; Wong, S.Y.-W.; Katyal, S.; Gao, Y.; McKinnon, P.J.; Lou, J.; Zhang, L.; Li, J.; Rebar, E.J.; et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011, 7, e1002080. [Google Scholar] [CrossRef] [PubMed]
- Boboila, C.; Jankovic, M.; Yan, C.T.; Wang, J.H.; Wesemann, D.R.; Zhang, T.; Fazeli, A.; Feldman, L.; Nussenzweig, A.; Nussenzweig, M.; et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc. Natl. Acad. Sci. USA 2010, 107, 3034–3039. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Siemann, M.; Grabos, M.; Murmann, T.; Pantelias, G.E.; Iliakis, G. Requirement for Parp-1 and DNA ligases 1 or 3 but not of Xrcc1 in chromosomal translocation formation by backup end joining. Nucleic Acids Res. 2014, 42, 6380–6392. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Piganeau, M.; Renouf, B.; Renaud, J.B.; Sallmyr, A.; Ruis, B.; Oh, S.; Tomkinson, A.E.; Hendrickson, E.A.; Giovannangeli, C.; et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 2014, 55, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Siemann, M.; Pantelias, G.E.; Iliakis, G. Marked contribution of alternative end-joining to chromosome translocation formation by stochastically induced DNA double strand breaks in G2-phase human cells. Mutat. Res. 2015, in press. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arakawa, H.; Iliakis, G. Alternative Okazaki Fragment Ligation Pathway by DNA Ligase III. Genes 2015, 6, 385-398. https://doi.org/10.3390/genes6020385
Arakawa H, Iliakis G. Alternative Okazaki Fragment Ligation Pathway by DNA Ligase III. Genes. 2015; 6(2):385-398. https://doi.org/10.3390/genes6020385
Chicago/Turabian StyleArakawa, Hiroshi, and George Iliakis. 2015. "Alternative Okazaki Fragment Ligation Pathway by DNA Ligase III" Genes 6, no. 2: 385-398. https://doi.org/10.3390/genes6020385
APA StyleArakawa, H., & Iliakis, G. (2015). Alternative Okazaki Fragment Ligation Pathway by DNA Ligase III. Genes, 6(2), 385-398. https://doi.org/10.3390/genes6020385