
Recovery from the DNA Replication Checkpoint


{kind=link}
Abstract
:1. Introduction
2. Activation of Checkpoint Signaling
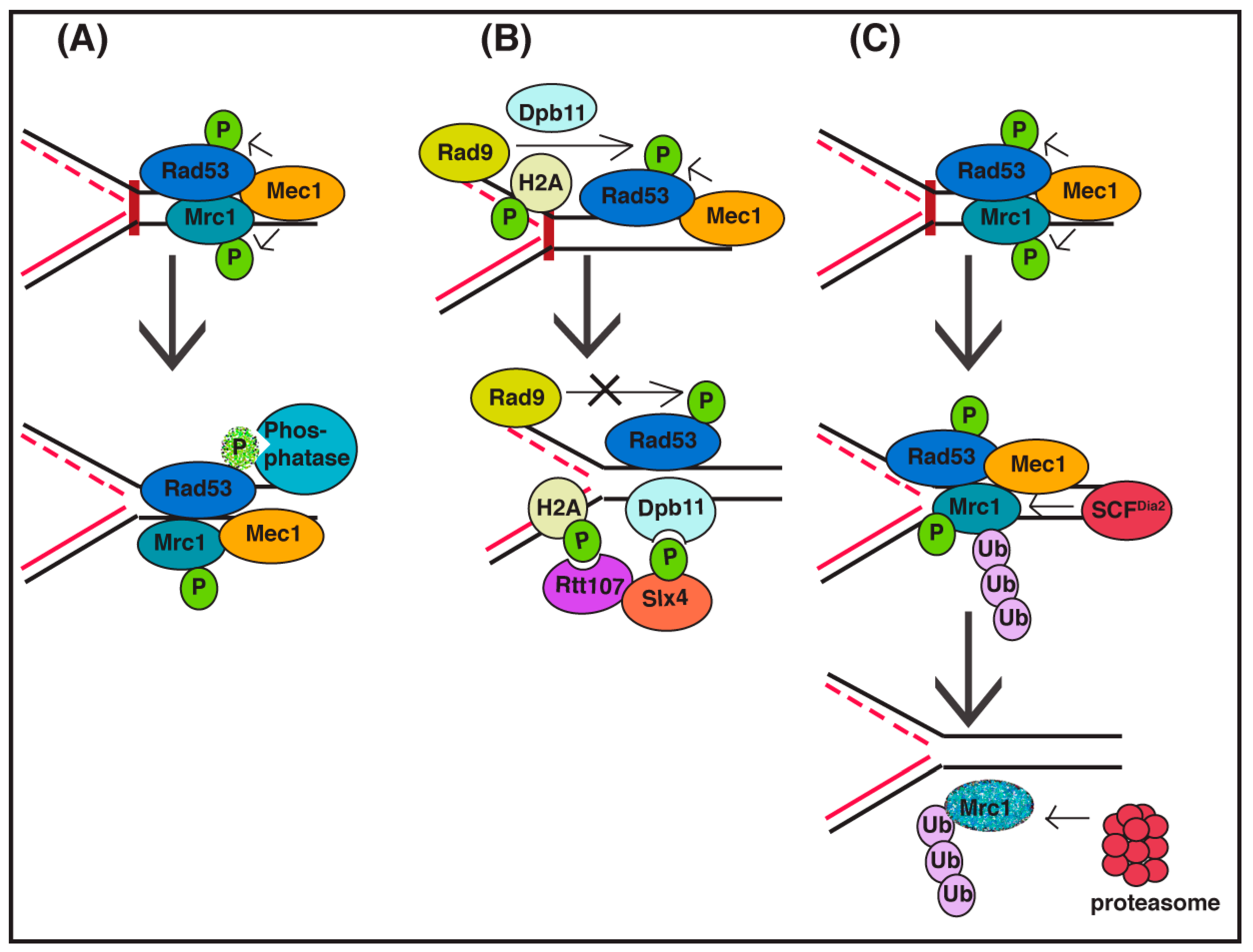
3. Checkpoint Signaling Inactivation
4. Resumption of DNA Replication
5. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations and Protein Name Derivations
| Rad | Radiation sensitive |
| Mec | Mitosis entry checkpoint |
| Ddc1 | DNA damage checkpoint 1 |
| ATR | ataxia telangiectasia mutated- and Rad3-related |
| Chk | Checkpoint kinase |
| Mrc1 | Mediator of replication checkpoint protein 1 |
| Csm3 | Chromosome segregation in meiosis protein 3 |
| Tof1 | Topoisomerase 1-associated factor 1 |
| Sgs1 | slow growth suppressor 1 |
| RecQ | recombination Q family |
| BLM | Bloom Syndrome protein |
| PP2A | protein phosphatase 2A |
| Pph3 | protein phosphatase 3 |
| Psy2 | platinum sensitivity 2 |
| PP2C | protein phosphatase 2C |
| Slx4 | Structure-Specific Endonuclease Subunit |
| Rtt | Regulator of Ty1 transposition protein |
| Ino80 | inositol requiring 80 |
| Isw2 | imitation switch 2 |
| Plk1 | Polo-like kinase 1 |
| SCF | Skp, Cullin, F-box protein containing complex |
| βTrCP | beta-transducin repeat protein |
| USP | ubiquitin-specific protease |
| HERC2 | HECT And RLD Domain Containing E3 Ubiquitin Protein Ligase 2 |
| BRCA1 | Breast cancer type 1 susceptibility protein |
| Dia2 | Digs into agar 2 |
| Mms22 | Methyl Methanesulfonate sensitivity 22 |
| METNASE | methyl transferase and nuclease |
| SETMAR | SET domain and mariner transposase fusion |
| HLTF | Helicase-like transcription factor |
| FANC | Fanconi Anemia group protein |
| Mph1 | mutator phenotype 1 |
| Fml1 | FANCM ortholog |
| SMARCAL | SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A-Like 1 |
| EEPD1 | Endonuclease/exonuclease/phosphatase family domain-containing protein 1 |
| WRN | Werner Syndrome protein |
References
- Lopes, M.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Plevani, P.; Muzi-Falconi, M.; Newlon, C.S.; Foiani, M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001, 412, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Katou, Y.; Kanoh, Y.; Bando, M.; Noguchi, H.; Tanaka, H.; Ashikari, T.; Sugimoto, K.; Shirahige, K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 2003, 424, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.B.; Zhou, Z.; Siede, W.; Friedberg, E.C.; Elledge, S.J. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 1994, 8, 2401–2415. [Google Scholar] [CrossRef] [PubMed]
- Alcasabas, A.A.; Osborn, A.J.; Bachant, J.; Hu, F.; Werler, P.J.; Bousset, K.; Furuya, K.; Diffley, J.F.; Carr, A.M.; Elledge, S.J. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 2001, 3, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Osborn, A.J.; Elledge, S.J. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003, 17, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Santocanale, C.; Diffley, J.F. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 1998, 395, 615–618. [Google Scholar] [PubMed]
- Shirahige, K.; Hori, Y.; Shiraishi, K.; Yamashita, M.; Takahashi, K.; Obuse, C.; Tsurimoto, T.; Yoshikawa, H. Regulation of DNA-replication origins during cell-cycle progression. Nature 1998, 395, 618–621. [Google Scholar] [PubMed]
- Paulovich, A.G.; Hartwell, L.H. A checkpoint regulates the rate of progression through S-phase in S. Cerevisiae in response to DNA damage. Cell 1995, 82, 841–847. [Google Scholar] [CrossRef]
- Boddy, M.N.; Russell, P. DNA replication checkpoint. Curr. Biol. 2001, 11, R953–R956. [Google Scholar] [CrossRef]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.C.; Haber, J.E. Surviving the breakup: The DNA damage checkpoint. Annu. Rev. Genet. 2006, 40, 209–235. [Google Scholar] [CrossRef] [PubMed]
- Majka, J.; Burgers, P.M. Yeast Rad17/Mec3/Ddc1: A sliding clamp for the DNA damage checkpoint. Proc. Natl. Acad. Sci. USA 2003, 100, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Venclovas, C.; Thelen, M.P. Structure-based predictions of Rad1, Rad9, Hus1 and Rad17 participation in sliding clamp and clamp-loading complexes. Nucleic Acids Res. 2000, 28, 2481–2493. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Desany, B.A.; Jones, W.J.; Liu, Q.; Wang, B.; Elledge, S.J. Regulation of Rad53 by the ATM-like kinases Mec1 and TEL1 in yeast cell cycle checkpoint pathways. Science 1996, 271, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fay, D.S.; Marini, F.; Foiani, M.; Stern, D.F. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev. 1996, 10, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kumagai, A.; Wang, S.X.; Dunphy, W.G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000, 14, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Hekmat-Nejad, M.; You, Z.; Yee, M.C.; Newport, J.W.; Cimprich, K.A. Xenopus Atr is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr. Biol. 2000, 10, 1565–1573. [Google Scholar] [CrossRef]
- Zhao, H.; Piwnica-Worms, H. Atr-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Foss, E.J. Tof1p regulates DNA damage responses during s phase in Saccharomyces cerevisiae. Genetics 2001, 157, 567–577. [Google Scholar] [PubMed]
- Tanaka, K.; Russell, P. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 2001, 3, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Bando, M.; Katou, Y.; Komata, M.; Tanaka, H.; Itoh, T.; Sutani, T.; Shirahige, K. Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J. Biol. Chem. 2009, 284, 34355–34365. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.A.; Bjergbaek, L.; Shimada, K.; Frei, C.; Gasser, S.M. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. Embo J. 2003, 22, 4325–4336. [Google Scholar] [CrossRef] [PubMed]
- Bjergbaek, L.; Cobb, J.A.; Tsai-Pflugfelder, M.; Gasser, S.M. Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. Embo J. 2005, 24, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Hegnauer, A.M.; Hustedt, N.; Shimada, K.; Pike, B.L.; Vogel, M.; Amsler, P.; Rubin, S.M.; van Leeuwen, F.; Guenole, A.; van Attikum, H.; et al. An N-terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. Embo J. 2012, 31, 3768–3783. [Google Scholar] [CrossRef] [PubMed]
- Szyjka, S.J.; Viggiani, C.J.; Aparicio, O.M. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol. Cell 2005, 19, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Tourriere, H.; Versini, G.; Cordon-Preciado, V.; Alabert, C.; Pasero, P. Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol. Cell 2005, 19, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Clemenson, C.; Marsolier-Kergoat, M.C. DNA damage checkpoint inactivation: Adaptation and recovery. DNA Repair 2009, 8, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Leroy, C.; Lee, S.E.; Vaze, M.B.; Ochsenbein, F.; Guerois, R.; Haber, J.E.; Marsolier-Kergoat, M.C. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol. Cell 2003, 11, 827–835, Erratum 2003, 11, 1119–1119. [Google Scholar]
- Guillemain, G.; Ma, E.; Mauger, S.; Miron, S.; Thai, R.; Guerois, R.; Ochsenbein, F.; Marsolier-Kergoat, M.C. Mechanisms of checkpoint kinase Rad53 inactivation after a double-strand break in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007, 27, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.M.; Szyjka, S.J.; Lis, E.T.; Bailey, A.O.; Yates, J.R.; Aparicio, O.M.; Romesberg, F.E. Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 9290–9295. [Google Scholar] [CrossRef] [PubMed]
- Travesa, A.; Duch, A.; Quintana, D.G. Distinct phosphatases mediate the deactivation of the DNA damage Checkpoint kinase Rad53. J. Biol. Chem. 2008, 283, 17123–17130. [Google Scholar] [CrossRef] [PubMed]
- Szyjka, S.J.; Aparicio, J.G.; Viggiani, C.J.; Knott, S.; Xu, W.; Tavare, S.; Aparicio, O.M. Rad53 regulates replication fork restart after DNA damage in Saccharomyces cerevisiae. Genes Dev. 2008, 22, 1906–1920. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Trastoy, M.; Berthonaud, V.; Chevalier, A.; Ducrot, C.; Marsolier-Kergoat, M.C.; Mann, C.; Leteurtre, F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene 2007, 26, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Liu, Y.; Ma, C.J.; Smolka, M.B. DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 2013, 493, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Balint, A.; Kim, T.; Gallo, D.; Cussiol, J.R.; Bastos de Oliveira, F.M.; Yimit, A.; Ou, J.; Nakato, R.; Gurevich, A.; Shirahige, K.; et al. Assembly of Slx4 signaling complexes behind DNA replication forks. Embo J. 2015, 34, 2182–2197. [Google Scholar] [CrossRef] [PubMed]
- Cussiol, J.R.; Jablonowski, C.M.; Yimit, A.; Brown, G.W.; Smolka, M.B. Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. Embo J. 2015, 34, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Dibitetto, D.; Ferrari, M.; Rawal, C.C.; Balint, A.; Kim, T.; Zhang, Z.; Smolka, M.B.; Brown, G.W.; Marini, F.; Pellicioli, A. Slx4 and Rtt107 control checkpoint signalling and DNA resection at double-strand breaks. Nucleic Acids Res. 2016, 44, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Jablonowski, C.M.; Cussiol, J.R.; Oberly, S.; Yimit, A.; Balint, A.; Kim, T.; Zhang, Z.; Brown, G.W.; Smolka, M.B. Termination of replication stress signaling via concerted action of the Slx4 scaffold and the PP4 phosphatase. Genetics 2015, 201, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Rodriguez, J.; Tsukiyama, T. Chromatin remodeling factors Isw2 and Ino80 regulate checkpoint activity and chromatin structure in S phase. Genetics 2015, 199, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Oma, Y.; Schleker, T.; Kugou, K.; Ohta, K.; Harata, M.; Gasser, S.M. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr. Biol. 2008, 18, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Peschiaroli, A.; Dorrello, N.V.; Guardavaccaro, D.; Venere, M.; Halazonetis, T.; Sherman, N.E.; Pagano, M. SCF beta TrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol. Cell 2006, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Bartek, J.; Lukas, J. Destruction of claspin by SCF beta TrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol. Cell 2006, 23, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.N.; Clarke, P.R. Regulation of claspin degradation by the ubiquitin-proteosome pathway during the cell cycle and in response to Atr-dependent checkpoint activation. FEBS Lett. 2006, 580, 4176–4181. [Google Scholar] [CrossRef] [PubMed]
- Mamely, I.; van Vugt, M.A.T.M.; Smits, V.A.J.; Semple, J.I.; Lemmens, B.; Perrakis, A.; Freire, R.; Medema, R.H. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr. Biol. 2006, 16, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zaugg, K.; Mak, T.W.; Elledge, S.J. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 2006, 126, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Faustrup, H.; Bekker-Jensen, S.; Bartek, J.; Lukas, J.; Mailand, N. USP7 counteracts SCFβTrCP- but not APCCdh1-mediated proteolysis of claspin. J. Cell. Biol. 2009, 184, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.; Cabrera, E.; Amoedo, H.; Hernandez-Perez, S.; Dominguez-Kelly, R.; Freire, R. USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination. Oncogene 2015, 34, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhao, H.; Liao, J.; Xu, X. HERC2/USP20 coordinates CHK1 activation by modulating CLASPIN stability. Nucleic Acids Res. 2014, 42, 13074–13081. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Deng, M.; Li, Y.; Yin, P.; Gao, B.; Fang, Y.; Wu, P.; Liu, T.; Lou, Z. HERC2-USP20 axis regulates DNA damage checkpoint through CLASPIN. Nucleic Acids Res. 2014, 42, 13110–13121. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Sundaramoorthy, E.; Rajendra, E.; Hattori, H.; Jeyasekharan, A.D.; Ayoub, N.; Schiess, R.; Aebersold, R.; Nishikawa, H.; Sedukhina, A.S.; et al. A DNA-damage selective role for BRCA1 E3 ligase in claspin ubiquitylation, CHK1 activation, and DNA repair. Curr. Biol. 2012, 22, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.M.; Arumugam, A.; Koepp, D.M. The Saccharomyces cerevisiae f-box protein Dia2 is a mediator of S-phase checkpoint recovery from DNA damage. Genetics 2013, 193, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Buser, R.; Kellner, V.; Melnik, A.; Wilson-Zbinden, C.; Schellhaas, R.; Kastner, L.; Piwko, W.; Dees, M.; Picotti, P.; Maric, M.; et al. The replisome-coupled E3 ubiquitin ligase Rtt101Mms22 counteracts Mrc1 function to tolerate genotoxic stress. PLoS Genet. 2016, 12, e1005843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tercero, J.A.; Diffley, J.F. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 2001, 412, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Tercero, J.A.; Longhese, M.P.; Diffley, J.F. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 2003, 11, 1323–1336. [Google Scholar] [CrossRef]
- Scorah, J.; McGowan, C.H. Claspin and Chk1 regulate replication fork stability by different mechanisms. Cell Cycle 2009, 8, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- De Piccoli, G.; Katou, Y.; Itoh, T.; Nakato, R.; Shirahige, K.; Labib, K. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol. Cell 2012, 45, 696–704. [Google Scholar] [CrossRef] [PubMed]
- De Haro, L.P.; Wray, J.; Williamson, E.A.; Durant, S.T.; Corwin, L.; Gentry, A.C.; Osheroff, N.; Lee, S.H.; Hromas, R.; Nickoloff, J.A. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 2010, 38, 5681–5691. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, M.; Williamson, E.; Nickoloff, J.; Lee, S.H.; Hromas, R. Metnase/setmar: A domesticated primate transposase that enhances DNA repair, replication, and decatenation. Genetica 2010, 138, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Hromas, R.; Williamson, E.A.; Fnu, S.; Lee, Y.J.; Park, S.J.; Beck, B.D.; You, J.S.; Leitao, A.; Nickoloff, J.A.; Lee, S.H. Chk1 phosphorylation of metnase enhances DNA repair but inhibits replication fork restart. Oncogene 2012, 31, 4245–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, E.A.; Wu, Y.; Singh, S.; Byrne, M.; Wray, J.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. The DNA repair component metnase regulates Chk1 stability. Cell Div. 2014, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.; Poli, J.; Marians, K.J.; Pasero, P. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013, 5, a012815. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Helleday, T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010, 11, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Blastyak, A.; Pinter, L.; Unk, I.; Prakash, L.; Prakash, S.; Haracska, L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell 2007, 28, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Achar, Y.J.; Balogh, D.; Haracska, L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc. Natl. Acad. Sci. USA 2011, 108, 14073–14078. [Google Scholar] [CrossRef] [PubMed]
- Gari, K.; Decaillet, C.; Stasiak, A.Z.; Stasiak, A.; Constantinou, A. The fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell 2008, 29, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.F.; Prakash, R.; Saro, D.; Longerich, S.; Niu, H.; Sung, P. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae mph1 protein. DNA Repair (Amst) 2011, 10, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Nandi, S.; Osman, F.; Ahn, J.S.; Jakovleska, J.; Lorenz, A.; Whitby, M.C. The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol. Cell 2008, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Betous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Bredemeyer, A.L.; Sowa, M.E.; Terret, M.E.; Jallepalli, P.V.; Harper, J.W.; Elledge, S.J. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009, 23, 2415–2425. [Google Scholar] [CrossRef] [PubMed]
- Bansbach, C.E.; Betous, R.; Lovejoy, C.A.; Glick, G.G.; Cortez, D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009, 23, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Yusufzai, T.; Kong, X.; Yokomori, K.; Kadonaga, J.T. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009, 23, 2400–2404. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ghosal, G.; Chen, J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009, 23, 2394–2399. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lee, S.H.; Williamson, E.A.; Reinert, B.L.; Cho, J.H.; Xia, F.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Brantley, A.; et al. EEPD1 rescues stressed replication forks and maintains genome stability by promoting end resection and homologous recombination repair. PLoS Genet. 2015, 11, e1005675. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different Rad51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Franchitto, A.; Pirzio, L.M.; Prosperi, E.; Sapora, O.; Bignami, M.; Pichierri, P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 2008, 183, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.L.; North, P.S.; Hickson, I.D. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 2007, 14, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J.M.; Li, N.; Folch, A.; Monnat, R.J., Jr. The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 2008, 7, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.B.; Hickson, I.D. RecQ helicases: Conserved guardians of genomic integrity. Adv. Exp. Med. Biol. 2013, 767, 161–184. [Google Scholar] [PubMed]
- Ashton, T.M.; Hickson, I.D. Yeast as a model system to study RecQ helicase function. DNA Repair (Amst) 2010, 9, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; D’Andrea, A.D. To the rescue: The Fanconi anemia genome stability pathway salvages replication forks. Cancer Cell 2012, 22, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Raghunandan, M.; Chaudhury, I.; Kelich, S.L.; Hanenberg, H.; Sobeck, A. FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi anemia core complex. Cell Cycle 2015, 14, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, I.; Sareen, A.; Raghunandan, M.; Sobeck, A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013, 41, 6444–6459. [Google Scholar] [CrossRef] [PubMed]
- Flott, S.; Alabert, C.; Toh, G.W.; Toth, R.; Sugawara, N.; Campbell, D.G.; Haber, J.E.; Pasero, P.; Rouse, J. Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol. Cell. Biol. 2007, 27, 6433–6445. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L.; Bergstralh, D.T.; Kohl, K.P.; LaRocque, J.R.; Moore, C.B.; Sekelsky, J. Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol. Cell 2009, 35, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Fekairi, S.; Scaglione, S.; Chahwan, C.; Taylor, E.R.; Tissier, A.; Coulon, S.; Dong, M.Q.; Ruse, C.; Yates, J.R., 3rd; Russell, P.; et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 2009, 138, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Munoz, I.M.; Hain, K.; Declais, A.C.; Gardiner, M.; Toh, G.W.; Sanchez-Pulido, L.; Heuckmann, J.M.; Toth, R.; Macartney, T.; Eppink, B.; et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 2009, 35, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, J.M.; Smogorzewska, A.; Sowa, M.E.; O’Connell, B.C.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 2009, 138, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.M.; Zaidi, I.W.; Vaisica, J.A.; Peter, M.; Brown, G.W. Regulation of Rtt107 recruitment to stalled DNA replication forks by the cullin Rtt101 and the Rtt109 acetyltransferase. Mol. Biol. Cell 2008, 19, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Almeida, B.S.; Smolka, M.B. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol. Cell 2010, 39, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.M.; Gohler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhury, I.; Koepp, D.M. Recovery from the DNA Replication Checkpoint. Genes 2016, 7, 94. https://doi.org/10.3390/genes7110094
Chaudhury I, Koepp DM. Recovery from the DNA Replication Checkpoint. Genes. 2016; 7(11):94. https://doi.org/10.3390/genes7110094
Chicago/Turabian StyleChaudhury, Indrajit, and Deanna M. Koepp. 2016. "Recovery from the DNA Replication Checkpoint" Genes 7, no. 11: 94. https://doi.org/10.3390/genes7110094
APA StyleChaudhury, I., & Koepp, D. M. (2016). Recovery from the DNA Replication Checkpoint. Genes, 7(11), 94. https://doi.org/10.3390/genes7110094