
Phylogenetic Relationships of the Fern Cyrtomium falcatum (Dryopteridaceae) from Dokdo Island Based on Chloroplast Genome Sequencing




Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Sequencing
2.2. Genome Analysis of the C. falcatum Chloroplast Genome
2.3. Comparative Chloroplast Genomic Analysis
2.4. Analysis of Single Sequence Repeats
2.5. Estimation of Substitution Rates
2.6. Analysis of RNA Editing
2.7. Phylogenetic Analysis
2.8. Molecular Clock Analysis
3. Results
3.1. General Characteristics of the Cyrtomium falcatum cp Genome
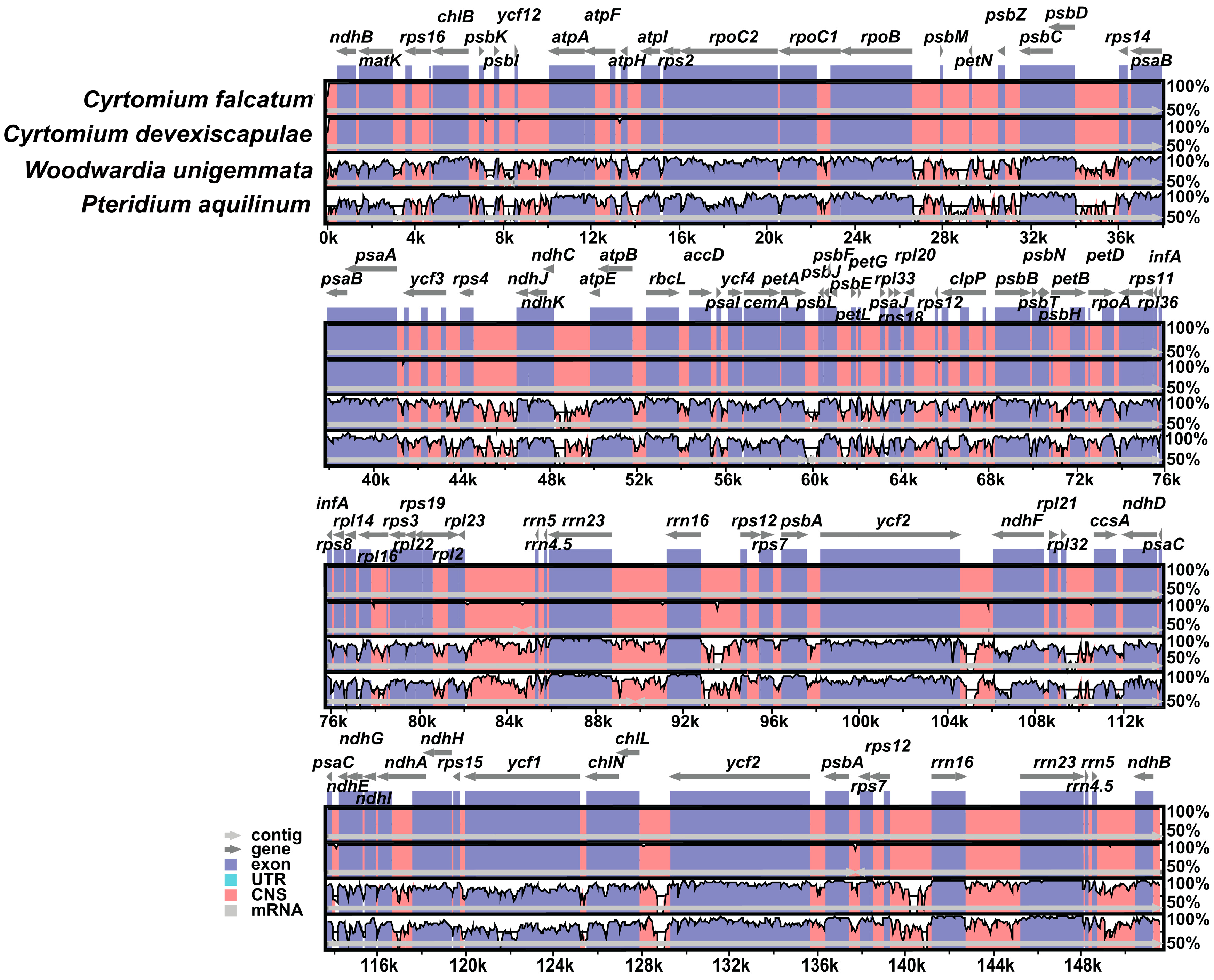
3.2. Comparative Analysis of Genome Structure
3.3. Repeat Sequence Analysis
3.4. Synonymous and Non-Synonymous Substitution Rate Analysis
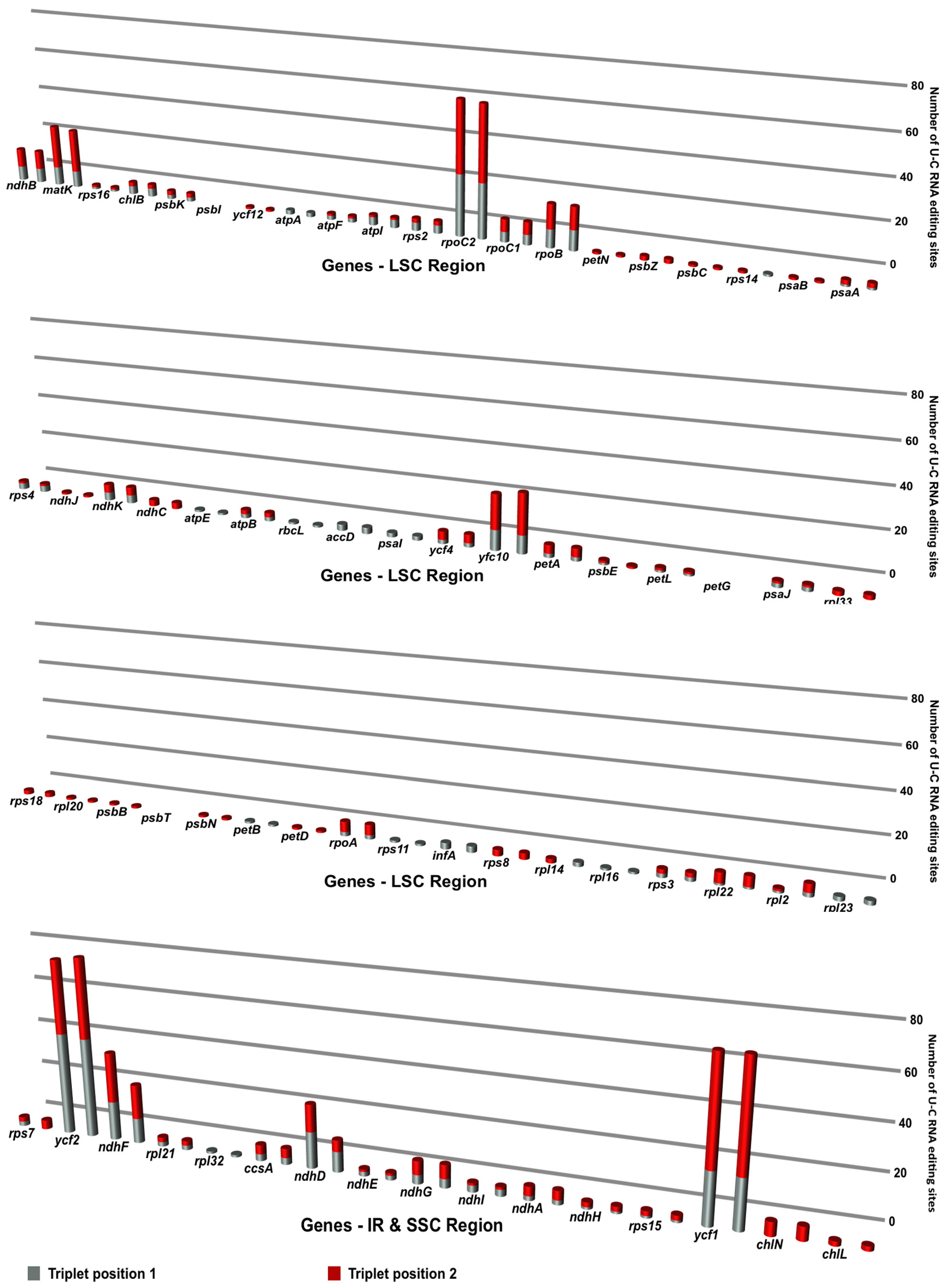
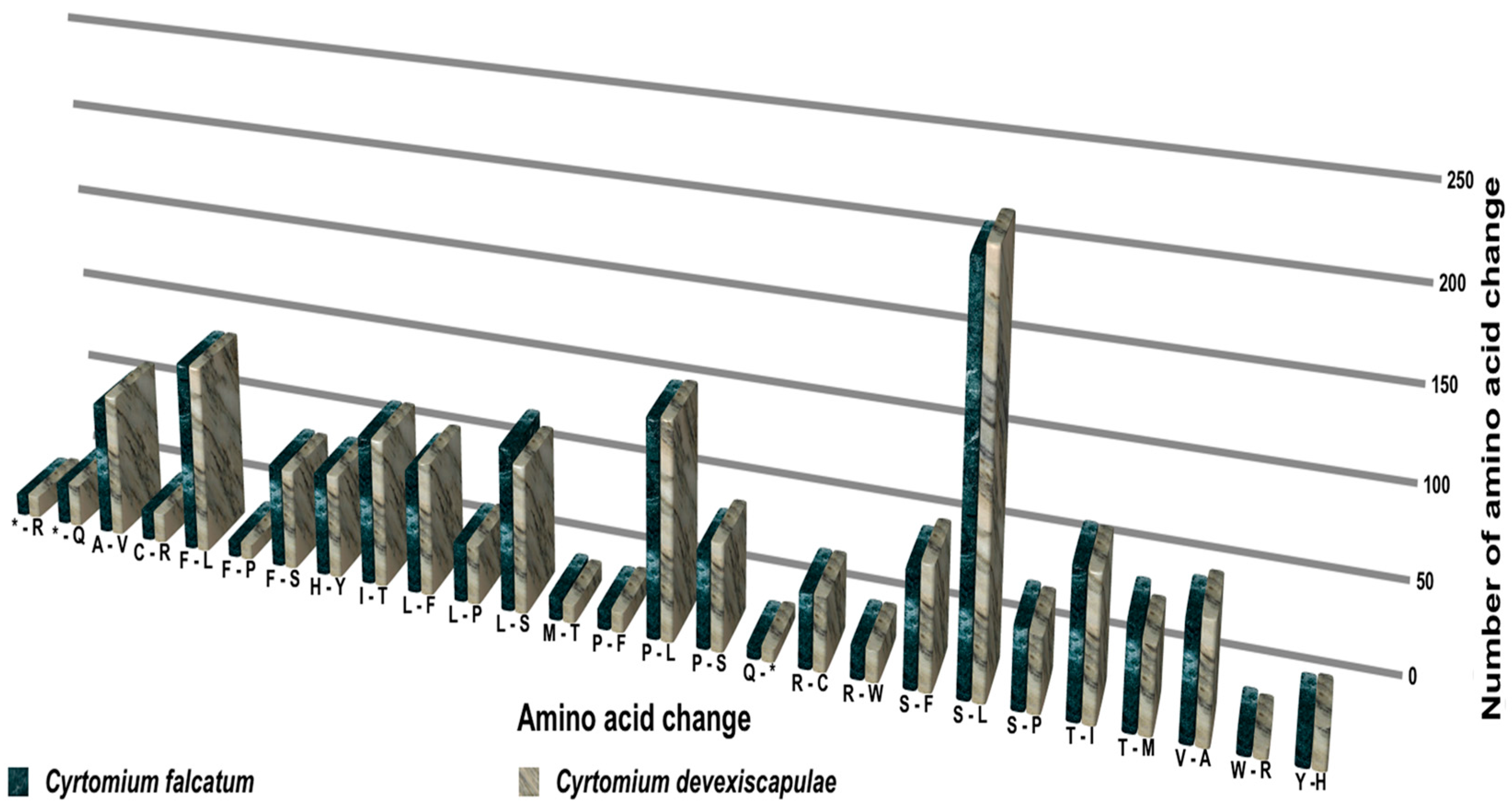
3.5. RNA Editing of Cyrtomium cp Genomes
3.6. Phylogenetic Analysis
3.7. Divergence Dating Analysis
4. Discussion
4.1. General Characteristics of the Cyrtomium falcatum cp Genome
4.2. Comparative Analysis of Genome Structure
4.3. Repeat Sequence Analysis
4.4. Synonymous and Non-Synonymous Substitution Rate Analysis
4.5. RNA Editing of Cyrtomium cp Genomes
4.6. Phylogenetic Analysis
4.7. Divergence Dating Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The complete nucleotide sequence of tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Hayashida, N.; Sugiura, M. Nicotiana chloroplast genes for components of the photosynthetic apparatus. Photosynth. Res. 1988, 18, 7–31. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Palmer, J.D. Use of chloroplast DNA rearrangements in reconstructing plant phylogeny. In Molecular Systematics of Plants; Soltis, P.S., Soltis, D.E., Doyle, J.J., Eds.; Chapman and Hall: New York, NY, USA, 1992; pp. 14–35. [Google Scholar]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Diversity and Evolution of Plants-Genotypic Variation in Higher Plants; Henry, R, Ed.; CABI Publishing: Wallingford, UK, 2005; pp. 45–68. [Google Scholar]
- Martin, G.; Baurens, F.C.; Cardi, C.; Aury, J.M.; D’Hont, A. The complete chloroplast genome of banana (Musa acuminata, Zingiberales): Insight into Plastid Monocotyledon Evolution. PLoS ONE 2013, 8, e67350. [Google Scholar] [CrossRef] [PubMed]
- Grewe, F.; Guo, W.; Gubbels, E.A.; Hansen, A.K.; Mower, J.P. Complete plastid genomes from Ophioglossum californicum, Psilotum nudum, and Equisetum hyemale reveal an ancestral land plant genome structure and resolve the position of Equisetales among monilophytes. BMC Evol. Biol. 2013, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yi, X.; Yang, Y.X.; Su, Y.J.; Wang, T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: Insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 2009, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Palmer, J.D. A chloroplast DNA inversion marks an ancient evolutionary split in the sunflower family (Asteraceae). Proc. Natl. Acad. Sci. USA 2009, 84, 5818–5822. [Google Scholar] [CrossRef]
- Graham, S.W.; Reeves, P.A.; Burns, A.C.E.; Olmstead, R.G. Microstructural changes in noncoding chloroplast DNA: Interpretation, evolution, and utility of indels and inversions in basal angiosperm phylogenetic inference. Int. J. Plant Sci. 2000, 161, S83–S96. [Google Scholar] [CrossRef]
- Lu, J.M.; Zhang, N.; Du, X.Y.; Wen, J.; Li, D.Z. Chloroplast phylogenomics resolves key relationships in ferns. J. Syst. Evol. 2015, 53, 448–457. [Google Scholar] [CrossRef]
- Pryer, K.M.; Schneider, H.; Smith, A.R.; Cranfill, R.; Wolf, P.G.; Hunt, J.S.; Sipes, S.D. Horsetails and ferns are a monophyletic group and the closest living relatives to seed plants. Nature 2001, 409, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.G.; Rowe, C.A.; Sinclair, R.B.; Hasebe, M. Complete nucleotide sequence of the chloroplast genome from a leptosporangiate fern, Adiantum capillus-veneris L. DNA Res. 2003, 10, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Franks, S.J. Genetics, evolution, and conservation of island plants. J. Plant Biol. 2010, 53, 1–9. [Google Scholar] [CrossRef]
- Jang, Y.D.; Park, B.J. Geology of Dokdo volcanic: Rocks, minerals, age, and cause of formation. In Nature of Dokdo; Research Institute for Ulleungdo & Dokdo Islands, Kyungpook National University, Eds.; Kyeongbuk University Press: Daegu, Korea, 2008; pp. 10–51. [Google Scholar]
- Kim, Y.K. Petrology of Ulleung volcanic island, Korea; Part 1. Geology. J. Jpn. Assoc. Miner. Pet. Econ. Geol. 1985, 80, 128–135. [Google Scholar] [CrossRef]
- The Korea Herald: [Weekender] Dokdo’s promising resources, ecology. Available online: http://www.koreaherald.com/view.php?ud=20140117000879 (accessed on 5 September 2016).
- Sun, B.Y.; Park, J.H.; Kwak, M.J. Characteristic of vascular flora of Ulleung and Dokdo. Rep. Surv. Nat. Environ. Korea 1996, 10, 113–135. [Google Scholar]
- Hyun, J.O.; Kwon, S.K. Flora of Dokdo. Report on the Detailed Survey of Dokdo Ecosystem; Ministry of Environment: Seoul, Korea, 2006; pp. 35–44.
- Song, G.; Park, S.J. Distribution and management of non-indigenous plants in Dokdo. Korean J. Plant Taxon. 2012, 42, 98–107. [Google Scholar] [CrossRef]
- Singh, A. Herbalism, Phytochemistry and Ethnopaharmacology; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Wyman, S.K.; Boore, J.L.; Jansen, R.K. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Der, J.P. Genomic Perspectives on Evolution in Bracken Fern. Ph.D. Thesis, Utah State University, Logan, UT, USA, 2010. [Google Scholar]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2009, 25, 1451–1452. [Google Scholar]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Leese, F.; Tollrian, R. Genome-wide analysis of tandem repeats in Daphnia pulex—A comparative approach. BMC Genom. 2010, 11, 277. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.; Knoop, V. PREPACT 2.0: Predicting C-to-U and U-to-C RNA editing in organelle genome sequences with multiple references and curated RNA editing annotation. Bioinform. Biol. Insights 2013, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.6. 2014. Available online: http://beast.bio.ed.ac.uk/Tracer.
- Pryer, K.M.; Schuettpelz, E.; Wolf, P.G.; Schneider, H.; Smith, A.R.; Cranfill, R. Phylogeny and evolution of ferns (monilophytes) with a focus on the early leptosporangiate divergences. Am. J. Bot. 2004, 91, 1582–1598. [Google Scholar] [CrossRef] [PubMed]
- Kong, W. The Geography of Dokdo; Ministry of Land, Infrastructure and Transport National Geographic Institute: Gyeonggi-do, Korea, 2011.
- Wolf, P.G.; Der, J.P.; Duffy, A.M.; Davidson, J.B.; Grusz, A.L.; Pryer, K.M. The evolution of chloroplast genes and genomes in ferns. Plant Mol. Biol. 2011, 7, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhou, Y.; Wang, Z.W.; Su, Y.J.; Wang, T. Evolution of the rpoB-psbZ region in fern plastid genomes: Notable structural rearrangements and highly variable intergenic spacers. BMC Plant Biol. 2011, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, C.; Sugita, M. Plastid transformation reveals that moss tRNAArg-CCG is not essential for plastid function. Plant J. 2004, 40, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983. [Google Scholar]
- Raman, G.; Park, S. Analysis of the Complete Chloroplast Genome of a Medicinal Plant, Dianthus superbus var. longicalyncinus, from a Comparative Genomics Perspective. PLoS ONE 2015, 10, e0141329. [Google Scholar] [CrossRef] [PubMed]
- Raman, G.; Park, S. The Complete Chloroplast Genome Sequence of Ampelopsis: Gene Organization, Comparative Analysis, and Phylogenetic Relationships to Other Angiosperms. Front. Plant Sci. 2016, 7, 341. [Google Scholar] [CrossRef] [PubMed]
- Makalowski, W.; Boguski, M.S. Evolutionary parameters of the transcribed mammalian genome: An analysis of 2820 orthologous rodent and human sequences. Proc. Natl. Acad. Sci. USA 1998, 95, 9407–9412. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Deng, L.; Jiang, Y.; Lu, P.; Yu, J. RNA editing sites exist in protein-coding genes in the chloroplast genome of Cycas taitungensis. J. Integr. Plant Biol. 2011, 53, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.B.; Goldschmidt-Clermont, M.; Hanson, M.R. Chloroplast RNA metabolism. Annu. Rev. Plant Biol. 2010, 61, 125–155. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Schuster, W. Evidence for a site-specific cytidine deamination reaction involved in C to U RNA editing of plant mitochondria. J. Biol. Chem. 1995, 270, 18227–18233. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Fan, H.; Suzuki, J.Y.; Wakasugi, T.; Tsudzuki, T.; Koessel, H.; Sugiura, M. Occurrence of silent RNA editing in chloroplasts: Its species specificity and the influence of environmental and developmental conditions. Plant Mol. Biol. 1996, 30, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Yura, K.; Go, M. Correlation between amino acid residues converted by RNA editing and functional residues in protein three-dimensional structures in plant organelles. BMC Plant Biol. 2008, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Mungpakdee, S.; Shinzato, C.; Takeuchi, T.; Kawashima, T.; Koyanagi, R.; Hisata, K.; Tanaka, M.; Goto, H.; Fujie, M.; Lin, S.; et al. Massive gene transfer and extensive RNA editing of a symbiotic dinoflagellate plastid genome. Genome Biol. Evol. 2014, 6, 1408–1422. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Pryer, K.M.; Schuettpelz, E.; Korall, P.; Schneider, H.; Wolf, P.G. A classification for extant ferns. Taxon 2006, 55, 705–731. [Google Scholar] [CrossRef]
- Gao, L.; Wang, B.; Wang, Z.W.; Zhou, Y.; Su, Y.J.; Wang, T. Plastome sequences of Lygodium japonicum and Marsilea crenata reveal the genome organization transformation from basal ferns to core leptosporangiates. Genome Biol. Evol. 2013, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.J.; Fong, R.; Collins, L.J.; McLenachan, P.A.; Penny, D. Two new fern chloroplasts and decelerated evolution linked to the long generation time in tree ferns. Genome Biol. Evol. 2014, 6, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Chung, M.G.; Kim, K.J. Chloroplast genome evolution in early diverged leptosporangiate ferns. Mol. Cells 2014, 37, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Des Marais, D.L.; Smith, A.R.; Britton, D.M.; Pryer, K.M. Phylogenetic relationships and evolution of extant horsetails, Equisetum, based on chloroplast DNA sequence data (rbcL and trnL-F). Int. J. Plant Sci. 2003, 164, 737–751. [Google Scholar] [CrossRef]
- Celaya, M.; McCabe, R. Kinematic model for the opening of the Sea of Japan and the bending of the Japanese islands. Geology 1987, 15, 53–57. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Group of Genes | Name of Genes | ||||
|---|---|---|---|---|---|---|
| RNA genes | Ribosomal RNA genes | rrn4.5 a | rrn5 a | rrn16 a | rrn23 a | |
| Transfer RNA genes | trnA-UGC a,b | trnC-GCA | trnD-GUC | trnE-UUC | trnF-GAA | |
| trnfM-CAU | trnG-GCC | trnG-UCC b | trnH-GUG a | trnI-CAU | ||
| trnI-GAU a,b | trnL-UAA b | trnL-UAG | trnM-CAU | trnN-GUU a | ||
| trnP-GGG | trnP-UGG | trnQ-UUG | trnR-ACG a | trnR-UCU | ||
| trnS-GCU | trnS-GGA | trnS-UGA | trnT-GGU | trnT-UGU a,b | ||
| trnV-UAC b | trnW-CCA | trnY-GUA | ||||
| Protein genes | Subunits of photosystem I | psaA | psaB | psaC | psaI | psaJ |
| ycf3 c | ycf4 | |||||
| Subunits of photosystem II | psbA a | psbB | psbC | psbD | psbE | |
| psbF | psbH | psbI | psbJ | psbK | ||
| psbL | psbM | psbN | psbT | psbZ | ||
| ycf12 | ||||||
| Subunits of cytochrome | petA | petB b | petD b | petG | petL | |
| petN | ||||||
| Subunits of ATP synthase | atpA | atpB | atpE | atpF b | atpH | |
| atpI | ||||||
| Large subunit of RuBisCO | rbcL | |||||
| Subunits of NADH dehydrogenase | ndhA b | ndhB a,b | ndhC | ndhD | ndhE | |
| ndhF | ndhG | ndhH | ndhI | ndhJ | ||
| ndhK | ||||||
| ATP-dependent protease subunit P | clpP c | |||||
| Chloroplast envelope membrane protein | cemA | |||||
| Light-independent Pchlide oxidoreductase (DPOR) | chlB | chlL | chlN | |||
| Ribosomal proteins | Small subunit of ribosome | rps2 | rps3 | rps4 | rps7 a | rps8 |
| rps11 | rps12 a,c,d | rps14 | rps15 | rps16 b | ||
| rps18 | rps19 | |||||
| Large subunit of ribosome | rpl2 | rpl14 | rpl16 b | rpl20 | rpl21 | |
| rpl22 | rpl23 | rpl32 | rpl33 | rpl36 | ||
| Transcription | DNA-dependent RNA polymerase | rpoA | rpoB | rpoC1 b | rpoC2 | |
| Translation | Translational initiation factor | infA | ||||
| Other proteins | Maturase | matK | ||||
| Subunit of acetyl-CoA | accD | |||||
| C-type cytochrome synthesis gene | ccsA | |||||
| Component of TIC complex | ycf1 | |||||
| Hypothetical proteins | ycf2 a | |||||
| Gene * | Location | Exon I | Intron I | Exon II | Intron II | Exon III |
|---|---|---|---|---|---|---|
| Nucleotides in Base Pairs | ||||||
| atpF | LSC | 145 | 720 | 410 | ||
| clpP | LSC | 240 | 571 7 | 292 | 723 | 71 |
| ndhA | SSC | 561 | 972 | 555 | ||
| ndhB | IR | 780 | 874 | 498 | ||
| petB | LSC | 6 | 848 | 642 | ||
| petD | LSC | 8 | 643 | 472 | ||
| rps12 # | LSC | 114 | -- | 232 | 577 | 26 |
| rpl2 | IR | 397 | 725 | 437 | ||
| rpl16 | LSC | 9 | 1068 | 399 | ||
| rpoC1 | LSC | 433 | 696 | 1610 | ||
| rps16 | LSC | 9 | 795 | 405 | ||
| trnA-UGC | IR | 36 | 798 | 37 | ||
| trnG-UCC | LSC | 23 | 907 | 48 | ||
| trnI-GAU | IR | 36 | 1016 | 36 | ||
| trnL-UAA | LSC | 34 | 637 | 51 | ||
| trnT-UGU | IR | 34 | 507 | 40 | ||
| trnV-UAC | LSC | 40 | 618 | 34 | ||
| ycf3 | LSC | 125 | 625 | 229 | 725 | 162 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raman, G.; Choi, K.S.; Park, S. Phylogenetic Relationships of the Fern Cyrtomium falcatum (Dryopteridaceae) from Dokdo Island Based on Chloroplast Genome Sequencing. Genes 2016, 7, 115. https://doi.org/10.3390/genes7120115
Raman G, Choi KS, Park S. Phylogenetic Relationships of the Fern Cyrtomium falcatum (Dryopteridaceae) from Dokdo Island Based on Chloroplast Genome Sequencing. Genes. 2016; 7(12):115. https://doi.org/10.3390/genes7120115
Chicago/Turabian StyleRaman, Gurusamy, Kyoung Su Choi, and SeonJoo Park. 2016. "Phylogenetic Relationships of the Fern Cyrtomium falcatum (Dryopteridaceae) from Dokdo Island Based on Chloroplast Genome Sequencing" Genes 7, no. 12: 115. https://doi.org/10.3390/genes7120115
APA StyleRaman, G., Choi, K. S., & Park, S. (2016). Phylogenetic Relationships of the Fern Cyrtomium falcatum (Dryopteridaceae) from Dokdo Island Based on Chloroplast Genome Sequencing. Genes, 7(12), 115. https://doi.org/10.3390/genes7120115