
Effects of Replication and Transcription on DNA Structure-Related Genetic Instability


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Repetitive Sequences, Non-B DNA Conformations and Genetic Instability
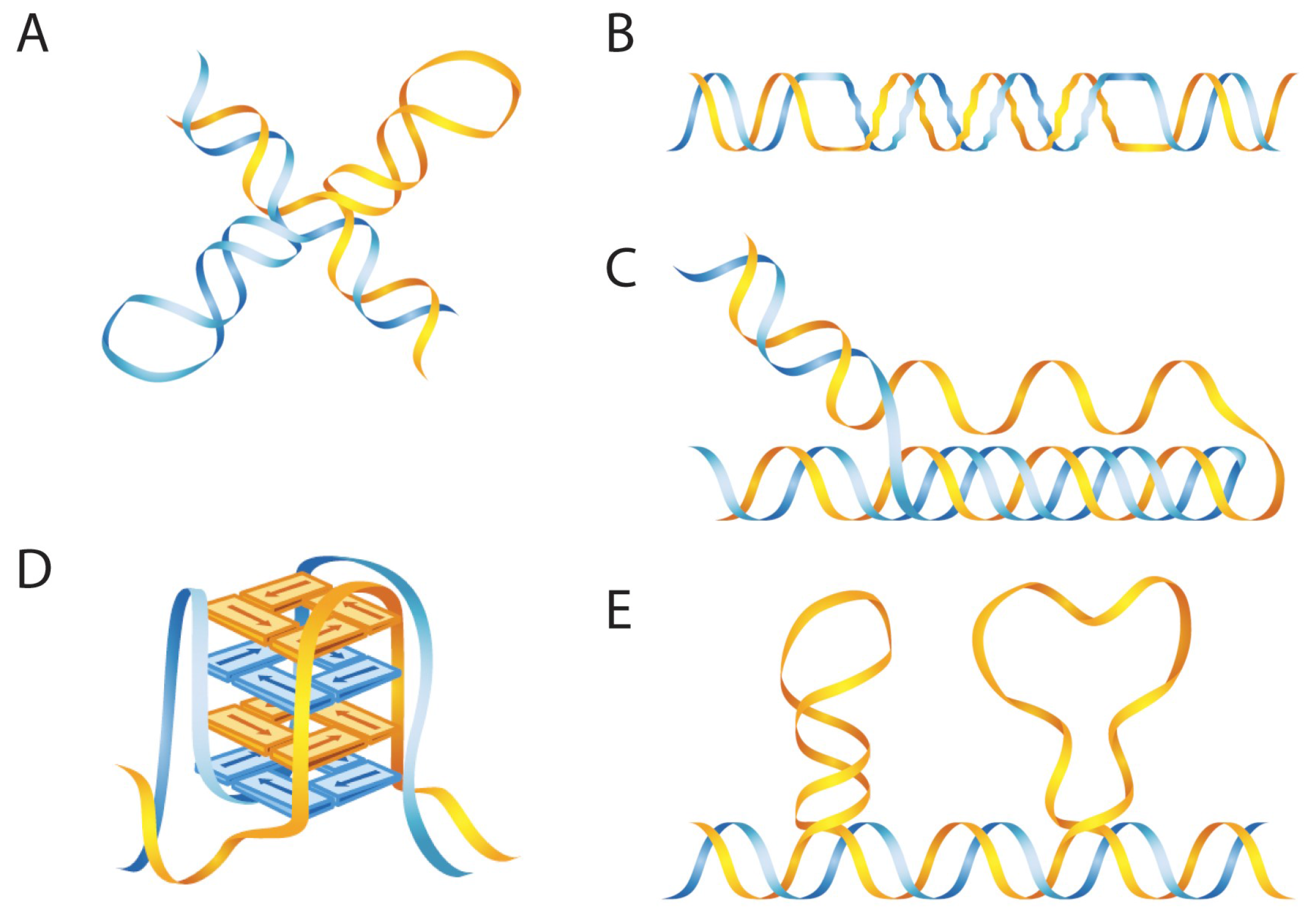
2.1. B-DNA and non-B DNA Structures
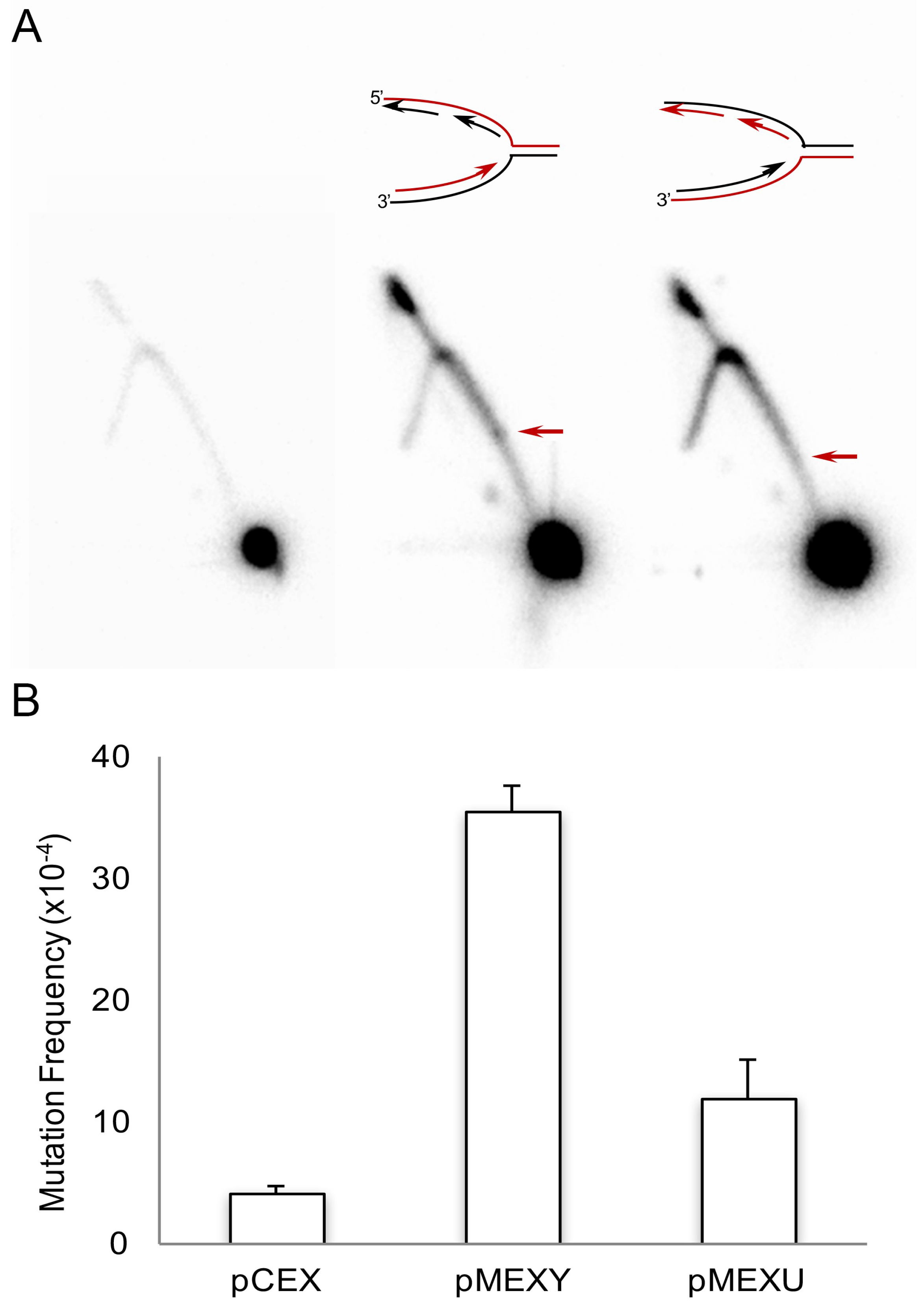
2.2. Non-B DNA-Forming Sequences Can Lead to Genetic Instability
3. DNA Replication and Transcription Facilitate Non-B DNA Structure Formation
3.1. DNA Replication and non-B DNA Formation
3.2. Transcription and non-B DNA formation
4. Non-B DNA Conformations Impact DNA Replication and Transcription
4.1. Non-B DNA Structures in front of Replication and Transcription machineries
4.2. Non-B DNA Structures behind Transcription Machinery
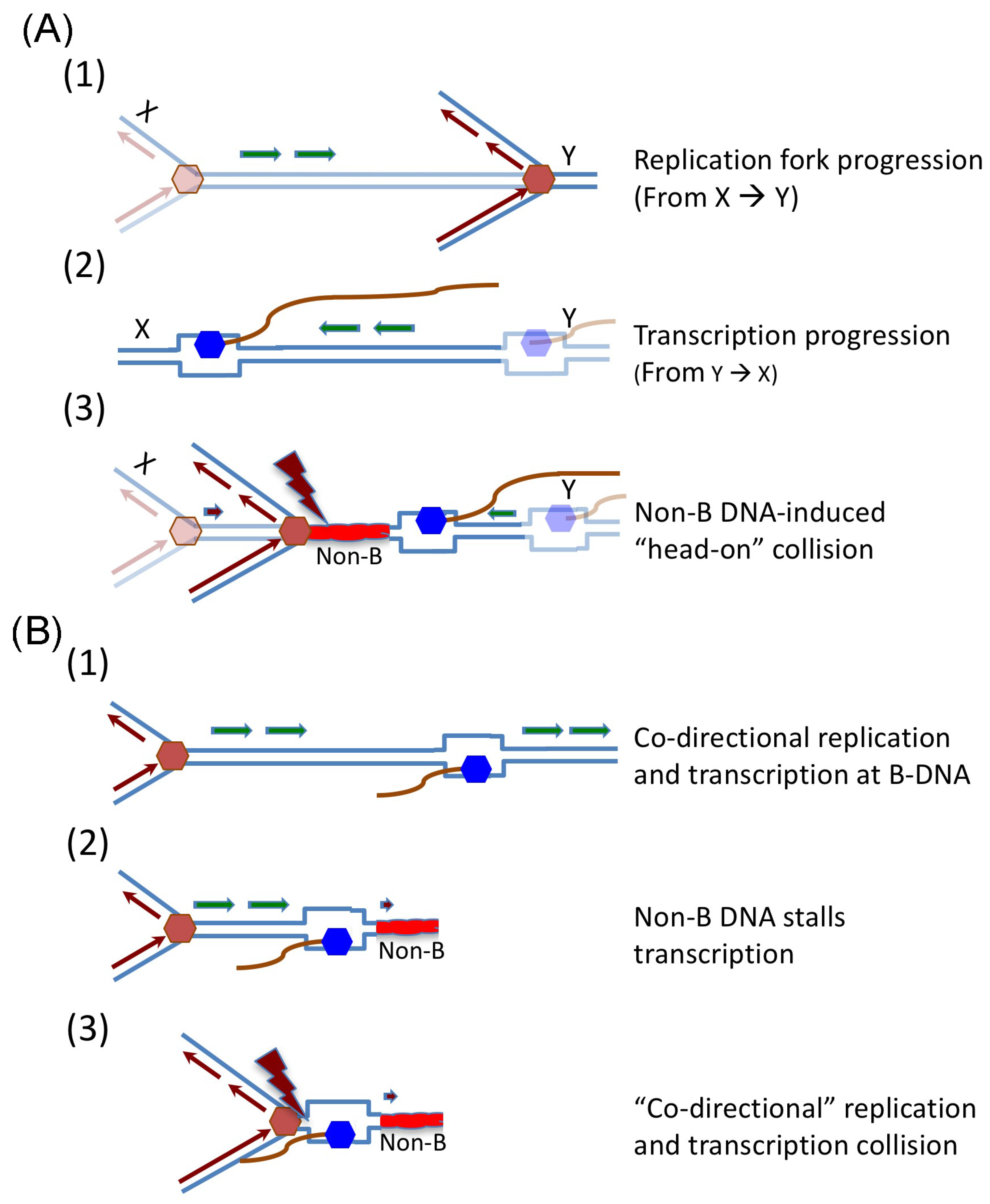
5. Non-B DNA Structures Cause Transcription and Replication Collision and Lead to Genetic Instability
6. Non-B DNA and Replication-Transcription Collision in Cancer
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, X.; Shen, Y.; Zhang, F.; Chiang, C.; Pillalamarri, V.; Blumenthal, I.; Talkowski, M.; Wu, B.L.; Gusella, J.F. Molecular analysis of a deletion hotspot in the NRXN1 region reveals the involvement of short inverted repeats in deletion CNVs. Am. J. Hum. Genet. 2013, 92, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Adair, G.M. Characterization of an apparent hotspot for spontaneous mutation in exon 5 of the Chinese hamster APRT gene. Mutat. Res. 1996, 352, 87–96. [Google Scholar] [CrossRef]
- De Graaff, E.; Rouillard, P.; Willems, P.J.; Smits, A.P.; Rousseau, F.; Oostra, B.A. Hotspot for deletions in the CGG repeat region of FMR1 in fragile X patients. Hum. Mol. Genet. 1995, 4, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, G.; Bacolla, A.; Zhao, J.; Spitser, S.; Vasquez, K.M. Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Rep. 2015, 10, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, M.C.; Piotrowski, A.; Alexander, J.; Callens, T.; Fu, C.; Mikhail, F.M.; Claes, K.B.; Messiaen, L. Palindrome-mediated and replication-dependent pathogenic structural rearrangements within the NF1 gene. Hum. Mutat. 2014, 35, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Majima, T. Conformational changes of non-B DNA. Chem. Soc. Rev. 2011, 40, 5893–5909. [Google Scholar] [CrossRef] [PubMed]
- Raynard, S.J.; Baker, M.D. Cis-acting regulatory sequences promote high-frequency gene conversion between repeated sequences in mammalian cells. Nucleic Acids Res. 2004, 32, 5916–5927. [Google Scholar] [CrossRef] [PubMed]
- Svetlova, E.Y.; Razin, S.V.; Debatisse, M. Mammalian recombination hot spot in a DNA loop anchorage region: A model for the study of common fragile sites. J. Cell. Biochem. 2001, 81, 170–178. [Google Scholar] [CrossRef]
- Waldman, A.S.; Tran, H.; Goldsmith, E.C.; Resnick, M.A. Long inverted repeats are an at-risk motif for recombination in mammalian cells. Genetics 1999, 153, 1873–1883. [Google Scholar] [PubMed]
- Hyrien, O.; Debatisse, M.; Buttin, G.; de Saint Vincent, B.R. A hotspot for novel amplification joints in a mosaic of Alu-like repeats and palindromic A + T-rich DNA. EMBO J. 1987, 6, 2401–2408. [Google Scholar] [PubMed]
- Watson, J.D.; Crick, F.H. The structure of DNA. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.D. Unusual DNA structures. J. Biol. Chem. 1988, 263, 1095–1098. [Google Scholar] [PubMed]
- Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability. Mutat. Res. 2006, 598, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Sinden, R.R.; Potaman, V.N.; Oussatcheva, E.A.; Pearson, C.E.; Lyubchenko, Y.L.; Shlyakhtenko, L.S. Triplet repeat DNA structures and human genetic disease: Dynamic mutations from dynamic DNA. J. Biosci. 2002, 27, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M.; Frank-Kamenetskii, M.D. H-DNA and related structures. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 541–576. [Google Scholar] [CrossRef] [PubMed]
- Htun, H.; Dahlberg, J.E. Topology and formation of triple-stranded H-DNA. Science 1989, 243, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Gilbert, W. A sodium-potassium switch in the formation of four-stranded G4-DNA. Nature 1990, 344, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Chaires, J.B.; Gray, R.D.; Trent, J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008, 36, 5482–5515. [Google Scholar] [CrossRef] [PubMed]
- Djian, P. Evolution of simple repeats in DNA and their relation to human disease. Cell 1998, 94, 155–160. [Google Scholar] [CrossRef]
- Malfoy, B.; Rousseau, N.; Vogt, N.; Viegas-Pequignot, E.; Dutrillaux, B.; Leng, M. Nucleotide sequence of an heterochromatic segment recognized by the antibodies to Z-DNA in fixed metaphase chromosomes. Nucleic Acids Res. 1986, 14, 3197–3214. [Google Scholar] [CrossRef] [PubMed]
- Johnston, B.H. Generation and detection of Z-DNA. Methods Enzymol. 1992, 211, 127–158. [Google Scholar] [PubMed]
- Wang, G.; Zhao, J.; Vasquez, K.M. Detection of cis- and trans-acting Factors in DNA Structure-Induced Genetic Instability Using In silico and Cellular Approaches. Front. Genet. 2016, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Baase, W.A.; Johnson, W.C., Jr. Circular dichroism and DNA secondary structure. Nucleic Acids Res. 1979, 6, 797–814. [Google Scholar] [CrossRef] [PubMed]
- Agazie, Y.M.; Lee, J.S.; Burkholder, G.D. Characterization of a new monoclonal antibody to triplex DNA and immunofluorescent staining of mammalian chromosomes. J. Biol. Chem. 1994, 269, 7019–7023. [Google Scholar] [PubMed]
- Raghavan, S.C.; Chastain, P.; Lee, J.S.; Hegde, B.G.; Houston, S.; Langen, R.; Hsieh, C.L.; Haworth, I.S.; Lieber, M.R. Evidence for a triplex DNA conformation at the bcl-2 major breakpoint region of the t(14;18) translocation. J. Biol. Chem. 2005, 280, 22749–22760. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Burkholder, G.D.; Latimer, L.J.; Haug, B.L.; Braun, R.P. A monoclonal antibody to triplex DNA binds to eucaryotic chromosomes. Nucleic Acids Res. 1987, 15, 1047–1061. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Latimer, L.J.; Haug, B.L.; Pulleyblank, D.E.; Skinner, D.M.; Burkholder, G.D. Triplex DNA in plasmids and chromosomes. Gene 1989, 82, 191–199. [Google Scholar] [CrossRef]
- Mikheikin, A.L.; Lushnikov, A.Y.; Lyubchenko, Y.L. Effect of DNA supercoiling on the geometry of holliday junctions. Biochemistry (Mosc.) 2006, 45, 12998–13006. [Google Scholar] [CrossRef] [PubMed]
- Shlyakhtenko, L.S.; Potaman, V.N.; Sinden, R.R.; Lyubchenko, Y.L. Structure and dynamics of supercoil-stabilized DNA cruciforms. J. Mol. Biol. 1998, 280, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, H.; Inagaki, H.; Yamada, K.; Ohye, T.; Taniguchi, M.; Emanuel, B.S.; Toda, T. Cruciform DNA structure underlies the etiology for palindrome-mediated human chromosomal translocations. J. Biol. Chem. 2004, 279, 35377–35383. [Google Scholar] [CrossRef] [PubMed]
- Vetcher, A.A.; Napierala, M.; Iyer, R.R.; Chastain, P.D.; Griffith, J.D.; Wells, R.D. Sticky DNA, a long GAA.GAA.TTC triplex that is formed intramolecularly, in the sequence of intron 1 of the frataxin gene. J. Biol. Chem. 2002, 277, 39217–39227. [Google Scholar] [CrossRef] [PubMed]
- Duquette, M.L.; Handa, P.; Vincent, J.A.; Taylor, A.F.; Maizels, N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004, 18, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.C.; Lieber, M.R. Chromosomal translocations and non-B DNA structures in the human genome. Cell Cycle 2004, 3, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Van Holde, K.; Zlatanova, J. Unusual DNA structures, chromatin and transcription. Bioessays 1994, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K.M. Models for chromosomal replication-independent non-B DNA structure-induced genetic instability. Mol. Carcinog. 2009, 48, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, H.; Ohye, T.; Kogo, H.; Kato, T.; Bolor, H.; Taniguchi, M.; Shaikh, T.H.; Emanuel, B.S.; Kurahashi, H. Chromosomal instability mediated by non-B DNA: Cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res. 2009, 19, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.; Pearson, C.E. Disease-associated repeat instability and mismatch repair. DNA Repair (Amst) 2016, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Lahue, R.S.; Slater, D.L. DNA repair and trinucleotide repeat instability. Front. Biosci. 2003, 8, s653–s665. [Google Scholar] [CrossRef] [PubMed]
- Bowater, R.P.; Wells, R.D. The intrinsically unstable life of DNA triplet repeats associated with human hereditary disorders. Prog. Nucleic Acid Res. Mol. Biol. 2001, 66, 159–202. [Google Scholar] [PubMed]
- Wells, R.D.; Dere, R.; Hebert, M.L.; Napierala, M.; Son, L.S. Advances in mechanisms of genetic instability related to hereditary neurological diseases. Nucleic Acids Res. 2005, 33, 3785–3798. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.G.; Todd, P.K. Repeat-associated non-AUG translation and its impact in neurodegenerative disease. Neurotherapeutics 2014, 11, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Wiener, F.; Ohno, S.; Babonits, M.; Sumegi, J.; Wirschubsky, Z.; Klein, G.; Mushinski, J.F.; Potter, M. Hemizygous interstitial deletion of chromosome 15 (band D) in three translocation-negative murine plasmacytomas. Proc. Natl. Acad. Sci. USA 1984, 81, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Akasaka, H.; Ueda, C.; Yonetani, N.; Maesako, Y.; Shimizu, A.; Yamabe, H.; Fukuhara, S.; Uchiyama, T.; Ohno, H. Molecular and clinical features of non-Burkitt’s, diffuse large-cell lymphoma of B-cell type associated with the c-MYC/immunoglobulin heavy-chain fusion gene. J. Clin. Oncol. 2000, 18, 510–518. [Google Scholar] [PubMed]
- Kovalchuk, A.L.; Muller, J.R.; Janz, S. Deletional remodeling of c-myc-deregulating chromosomal translocations. Oncogene 1997, 15, 2369–2377. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Haluska, F.G.; Falk, M.H.; Henglein, B.; Hameister, H.; Croce, C.M.; Bornkamm, G.W. Mapping chromosomal breakpoints of Burkitt’s t(8;14) translocations far upstream of c-myc. Cancer Res. 1992, 52, 6547–6552. [Google Scholar] [PubMed]
- Haluska, F.G.; Tsujimoto, Y.; Croce, C.M. The t(8;14) breakpoint of the EW 36 undifferentiated lymphoma cell line lies 5′ of MYC in a region prone to involvement in endemic Burkitt’s lymphomas. Nucleic Acids Res. 1988, 16, 2077–2085. [Google Scholar] [CrossRef] [PubMed]
- Saglio, G.; Grazia Borrello, M.; Guerrasio, A.; Sozzi, G.; Serra, A.; di Celle, P.F.; Foa, R.; Ferrarini, M.; Roncella, S.; Borgna Pignatti, C.; et al. Preferential clustering of chromosomal breakpoints in Burkitt’s lymphomas and L3 type acute lymphoblastic leukemias with a t(8;14) translocation. Genes Chromosomes Cancer 1993, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Care, A.; Cianetti, L.; Giampaolo, A.; Sposi, N.M.; Zappavigna, V.; Mavilio, F.; Alimena, G.; Amadori, S.; Mandelli, F.; Peschle, C. Translocation of c-myc into the immunoglobulin heavy-chain locus in human acute B-cell leukemia. A molecular analysis. EMBO J. 1986, 5, 905–911. [Google Scholar] [PubMed]
- Wilda, M.; Busch, K.; Klose, I.; Keller, T.; Woessmann, W.; Kreuder, J.; Harbott, J.; Borkhardt, A. Level of MYC overexpression in pediatric Burkitt’s lymphoma is strongly dependent on genomic breakpoint location within the MYC locus. Genes Chromosomes Cancer 2004, 41, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Rimokh, R.; Rouault, J.P.; Wahbi, K.; Gadoux, M.; Lafage, M.; Archimbaud, E.; Charrin, C.; Gentilhomme, O.; Germain, D.; Samarut, J.; et al. A chromosome 12 coding region is juxtaposed to the MYC protooncogene locus in a t(8;12)(q24;q22) translocation in a case of B-cell chronic lymphocytic leukemia. Genes Chromosomes Cancer 1991, 3, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Wolfl, S.; Wittig, B.; Rich, A. Identification of transcriptionally induced Z-DNA segments in the human c-myc gene. Biochim. Biophys. Acta 1995, 1264, 294–302. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Grand, C.L.; Han, H.; Munoz, R.M.; Weitman, S.; Von Hoff, D.D.; Hurley, L.H.; Bearss, D.J. The cationic porphyrin TMPyP4 down-regulates c-MYC and human telomerase reverse transcriptase expression and inhibits tumor growth in vivo. Mol. Cancer Ther. 2002, 1, 565–573. [Google Scholar] [PubMed]
- Juranek, S.A.; Paeschke, K. Cell cycle regulation of G-quadruplex DNA structures at telomeres. Curr. Pharm. Des. 2012, 18, 1867–1872. [Google Scholar] [CrossRef] [PubMed]
- Capra, J.A.; Paeschke, K.; Singh, M.; Zakian, V.A. G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput. Biol. 2010, 6, e1000861. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, H.; Inagaki, H.; Ohye, T.; Kogo, H.; Kato, T.; Emanuel, B.S. Palindrome-mediated chromosomal translocations in humans. DNA Repair (Amst) 2006, 5, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Tainer, J.A.; Vasquez, K.M.; Cooper, D.N. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 2016, 44, 5673–5688. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K.M. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 13448–13453. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Carbajal, S.; Vijg, J.; DiGiovanni, J.; Vasquez, K.M. DNA structure-induced genomic instability in vivo. J. Natl. Cancer Inst. 2008, 100, 1815–1817. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Christensen, L.A.; Vasquez, K.M. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2677–2682. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Gaddis, S.; Vasquez, K.M. Methods to detect replication-dependent and replication-independent DNA structure-induced genetic instability. Methods 2013, 64, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Caskey, C.T.; Pizzuti, A.; Fu, Y.H.; Fenwick, R.G., Jr.; Nelson, D.L. Triplet repeat mutations in human disease. Science 1992, 256, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Martorell, L.; Monckton, D.G.; Gamez, J.; Johnson, K.J.; Gich, I.; Lopez de Munain, A.; Baiget, M. Progression of somatic CTG repeat length heterogeneity in the blood cells of myotonic dystrophy patients. Hum. Mol. Genet. 1998, 7, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.J.; Ashizawa, T.; Monckton, D.G.; Caskey, C.T.; Richards, C.S. Somatic heterogeneity of the CTG repeat in myotonic dystrophy is age and size dependent. Am. J. Hum. Genet. 1995, 56, 114–122. [Google Scholar] [PubMed]
- Martorell, L.; Martinez, J.M.; Carey, N.; Johnson, K.; Baiget, M. Comparison of CTG repeat length expansion and clinical progression of myotonic dystrophy over a five year period. J. Med. Genet. 1995, 32, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Wohrle, D.; Kennerknecht, I.; Wolf, M.; Enders, H.; Schwemmle, S.; Steinbach, P. Heterogeneity of DM kinase repeat expansion in different fetal tissues and further expansion during cell proliferation in vitro: Evidence for a casual involvement of methyl-directed DNA mismatch repair in triplet repeat stability. Hum. Mol. Genet. 1995, 4, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M.; Smirnova, E.V. Positioned to expand. Nat. Genet. 2002, 31, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.K.; Yin, Y.; Petes, T.D.; Symington, L.S. Mre11-Sae2 and RPA Collaborate to Prevent Palindromic Gene Amplification. Mol. Cell 2015, 60, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Audry, J.; Maestroni, L.; Delagoutte, E.; Gauthier, T.; Nakamura, T.M.; Gachet, Y.; Saintomé, C.; Géli, V.; Coulon, S. RPA prevents G-rich structure formation at lagging-strand telomeres to allow maintenance of chromosome ends. EMBO J. 2015, 34, 1942–1958. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.K.; Chen, H.; Symington, L.S. Replication protein A prevents promiscuous annealing between short sequence homologies: Implications for genome integrity. Bioessays 2015, 37, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Vlijm, R.; Mashaghi, A.; Bernard, S.; Modesti, M.; Dekker, C. Experimental phase diagram of negatively supercoiled DNA measured by magnetic tweezers and fluorescence. Nanoscale 2015, 7, 3205–3216. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.; Sokoloski, J.; Galletto, R.; Elson, E.L.; Wold, M.S.; Lohman, T.M. Diffusion of human replication protein A along single-stranded DNA. J. Mol. Biol. 2014, 426, 3246–3261. [Google Scholar] [CrossRef] [PubMed]
- Safa, L.; Delagoutte, E.; Petruseva, I.; Alberti, P.; Lavrik, O.; Riou, J.F.; Saintomé, C. Binding polarity of RPA to telomeric sequences and influence of G-quadruplex stability. Biochimie 2014, 103, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, R.; Inoue, J.; Mikawa, T.; Ha, T. Single molecule analysis of Thermus thermophilus SSB protein dynamics on single-stranded DNA. Nucleic Acids Res. 2014, 42, 3821–3832. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Jaworski, A.; Ohshima, K.; Wells, R.D. Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat. Genet. 1995, 10, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Miret, J.J.; Pessoa-Brandao, L.; Lahue, R.S. Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotide repeats in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1998, 95, 12438–12443. [Google Scholar] [CrossRef] [PubMed]
- Samadashwily, G.M.; Raca, G.; Mirkin, S.M. Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 1997, 17, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.Q.; Sinden, R.R. Preferential DNA secondary structure mutagenesis in the lagging strand of replication in E. coli. Nature 1991, 352, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Hashem, V.I.; Sinden, R.R. Duplications between direct repeats stabilized by DNA secondary structure occur preferentially in the leading strand during DNA replication. Mutat. Res. 2005, 570, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.R.; Wells, R.D. Expansion and deletion of triplet repeat sequences in Escherichia coli occur on the leading strand of DNA replication. J. Biol. Chem. 1999, 274, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, R.; Krasilnikova, M.M.; Samadashwily, G.M.; Lahue, R.; Mirkin, S.M. Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 2003, 23, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Chen, X.; Bissler, J.J.; Sinden, R.R.; Leffak, M. Replication-dependent instability at (CTG) x (CAG) repeat hairpins in human cells. Nat. Chem. Biol. 2010, 6, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Maurer, D.J.; O’Callaghan, B.L.; Livingston, D.M. Orientation dependence of trinucleotide CAG repeat instability in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 6617–6622. [Google Scholar] [CrossRef] [PubMed]
- Freudenreich, C.H.; Stavenhagen, J.B.; Zakian, V.A. Stability of a CTG/CAG trinucleotide repeat in yeast is dependent on its orientation in the genome. Mol. Cell. Biol. 1997, 17, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Hartenstine, M.J.; Goodman, M.F.; Petruska, J. Base stacking and even/odd behavior of hairpin loops in DNA triplet repeat slippage and expansion with DNA polymerase. J. Biol. Chem. 2000, 275, 18382–18390. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.D.; Nichol, K.; Wang, Y.H.; Pearson, C.E. Evidence of cis-acting factors in replication-mediated trinucleotide repeat instability in primate cells. Nat. Genet. 2002, 31, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.A.; Shishkin, A.A.; Voineagu, I.; Pavlov, Y.I.; Shcherbakova, P.V.; Mirkin, S.M. Role of DNA polymerases in repeat-mediated genome instability. Cell Rep. 2012, 2, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.S.; Chang, H.C.; Boudi, F.B.; Reddy, S. CTG repeats show bimodal amplification in E. coli. Cell 1998, 95, 531–540. [Google Scholar] [CrossRef]
- Spiro, C.; Pelletier, R.; Rolfsmeier, M.L.; Dixon, M.J.; Lahue, R.S.; Gupta, G.; Park, M.S.; Chen, X.; Mariappan, S.V.; McMurray, C.T. Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol. Cell 1999, 4, 1079–1085. [Google Scholar] [CrossRef]
- Iyer, R.R.; Pluciennik, A.; Rosche, W.A.; Sinden, R.R.; Wells, R.D. DNA polymerase III proofreading mutants enhance the expansion and deletion of triplet repeat sequences in Escherichia coli. J. Biol. Chem. 2000, 275, 2174–2184. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jinks-Robertson, S. Transcription as a source of genome instability. Nat. Rev. Genet. 2012, 13, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Wittig, B.; Dorbic, T.; Rich, A. Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei. Proc. Natl. Acad. Sci. USA 1991, 88, 2259–2263. [Google Scholar] [CrossRef] [PubMed]
- Wittig, B.; Wolfl, S.; Dorbic, T.; Vahrson, W.; Rich, A. Transcription of human c-myc in permeabilized nuclei is associated with formation of Z-DNA in three discrete regions of the gene. EMBO J. 1992, 11, 4653–4663. [Google Scholar] [PubMed]
- Cerna, A.; Cuadrado, A.; Jouve, N.; Diaz de la Espina, S.M.; De la Torre, C. Z-DNA, a new in situ marker for transcription. Eur. J. Histochem. 2004, 48, 49–56. [Google Scholar] [PubMed]
- Lombrana, R.; Almeida, R.; Alvarez, A.; Gomez, M. R-loops and initiation of DNA replication in human cells: A missing link? Front. Genet. 2015, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Peleg, M.; Kopel, V.; Borowiec, J.A.; Manor, H. Formation of DNA triple helices inhibits DNA unwinding by the SV40 large T-antigen helicase. Nucleic Acids Res. 1995, 23, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Kopel, V.; Pozner, A.; Baran, N.; Manor, H. Unwinding of the third strand of a DNA triple helix, a novel activity of the SV40 large T-antigen helicase. Nucleic Acids Res. 1996, 24, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Hoyne, P.R.; Maher, L.J., III. Functional studies of potential intrastrand triplex elements in the Escherichia coli genome. J. Mol. Biol. 2002, 318, 373–386. [Google Scholar] [CrossRef]
- Hile, S.E.; Eckert, K.A. Positive correlation between DNA polymerase alpha-primase pausing and mutagenesis within polypyrimidine/polypurine microsatellite sequences. J. Mol. Biol. 2004, 335, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Rao, B.S. Pausing of simian virus 40 DNA replication fork movement in vivo by (dG-dA)n.(dT-dC)n tracts. Gene 1994, 140, 233–237. [Google Scholar] [CrossRef]
- Krasilnikova, M.M.; Mirkin, S.M. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol. Cell. Biol. 2004, 24, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Narayanan, V.; Lobachev, K.S.; Mirkin, S.M. Replication stalling at unstable inverted repeats: Interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 9936–9941. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.P.; Shah, K.A.; Niu, H.; Sung, P.; Mirkin, S.M.; Freudenreich, C.H. Overcoming natural replication barriers: Differential helicase requirements. Nucleic Acids Res. 2012, 40, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Surka, C.F.; Shishkin, A.A.; Krasilnikova, M.M.; Mirkin, S.M. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 2009, 16, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, J.Q.; Chen, Z.; Zheng, K.W.; Chen, C.Y.; Hao, Y.H.; Tan, Z. G-quadruplex formation at the 3′ end of telomere DNA inhibits its extension by telomerase, polymerase and unwinding by helicase. Nucleic Acids Res. 2011, 39, 6229–6237. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Hurley, L.H.; Salazar, M. A DNA polymerase stop assay for G-quadruplex-interactive compounds. Nucleic Acids Res. 1999, 27, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Paeschke, K.; Capra, J.A.; Zakian, V.A. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 2011, 145, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, J.; Vasquez, K.M. Methods to determine DNA structural alterations and genetic instability. Methods 2009, 48, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Reines, D.; Hanawalt, P.C. Structural characterization of RNA polymerase II complexes arrested by a cyclobutane pyrimidine dimer in the transcribed strand of template DNA. J. Biol. Chem. 1999, 274, 24124–24130. [Google Scholar] [CrossRef] [PubMed]
- Ditlevson, J.V.; Tornaletti, S.; Belotserkovskii, B.P.; Teijeiro, V.; Wang, G.; Vasquez, K.M.; Hanawalt, P.C. Inhibitory effect of a short Z-DNA forming sequence on transcription elongation by T7 RNA polymerase. Nucleic Acids Res. 2008, 36, 3163–3170. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; De Silva, E.; Tornaletti, S.; Wang, G.; Vasquez, K.M.; Hanawalt, P.C. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J. Biol. Chem. 2007, 282, 32433–32441. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Ogloblina, A.M.; Belotserkovskii, B.P.; Dolinnaya, N.G.; Yakubovskaya, M.G.; Mirkin, S.M.; Hanawalt, P.C. Transcription blockage by stable H-DNA analogs in vitro. Nucleic Acids Res. 2015, 43, 6994–7004. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; Liu, R.; Tornaletti, S.; Krasilnikova, M.M.; Mirkin, S.M.; Hanawalt, P.C. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc. Natl. Acad. Sci. USA 2010, 107, 12816–12821. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Mirkin, S.M. Mechanisms of transcription-replication collisions in bacteria. Mol. Cell. Biol. 2005, 25, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Olavarrieta, L.; Martinez-Robles, M.L.; Hernandez, P.; Krimer, D.B.; Schvartzman, J.B. Knotting dynamics during DNA replication. Mol. Microbiol. 2002, 46, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Jinks-Robertson, S. dUTP incorporation into genomic DNA is linked to transcription in yeast. Nature 2009, 459, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.; Wang, Y.H. DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genom. 2015, 16, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Debacker, K.; Kooy, R.F. Fragile sites and human disease. Hum. Mol. Genet. 2007, 16, R150–R158. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Durkin, S.G.; Ragland, R.L.; Glover, T.W. Common fragile sites as targets for chromosome rearrangements. DNA Repair (Amst) 2006, 5, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.R. Rare fragile sites. Cytogenet. Genome Res. 2003, 100, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W. Instability at chromosomal fragile sites. Recent Results Cancer Res. 1998, 154, 185–199. [Google Scholar] [PubMed]
- Ohta, M.; Inoue, H.; Cotticelli, M.G.; Kastury, K.; Baffa, R.; Palazzo, J.; Siprashvili, Z.; Mori, M.; McCue, P.; Druck, T.; et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996, 84, 587–597. [Google Scholar] [CrossRef]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.I.; Zhu, Y.; McAvoy, S.; Kuhn, R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006, 232, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013, 4, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callen, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Brambati, A.; Colosio, A.; Zardoni, L.; Galanti, L.; Liberi, G. Replication and transcription on a collision course: Eukaryotic regulation mechanisms and implications for DNA stability. Front Genet 2015, 6, 166. [Google Scholar] [CrossRef] [PubMed]
- Sankar, T.S.; Wastuwidyaningtyas, B.D.; Dong, Y.; Lewis, S.A.; Wang, J.D. The nature of mutations induced by replication-transcription collisions. Nature 2016, 535, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Herkert, B.; Eilers, M. Facilitating replication under stress: An oncogenic function of MYC? Nat. Rev. Cancer 2009, 9, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Shigemi, Z.; Baba, Y.; Hara, N.; Matsuhiro, J.; Kagawa, H.; Watanabe, T.; Fujimuro, M. Effects of ER stress on unfolded protein responses, cell survival, and viral replication in primary effusion lymphoma. Biochem. Biophys. Res. Commun. 2016, 469, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Barone, G.; Staples, C.J.; Ganesh, A.; Patterson, K.W.; Bryne, D.P.; Myers, K.N.; Patil, AA.; Eyers, C.E.; Maslen, S.; Skehel, J.M.; et al. Human CDK18 promotes replication stress signaling and genome stability. Nucleic Acids Res. 2016, 44, 8772–8785. [Google Scholar] [CrossRef] [PubMed]
- Palou, G.; Palou, R.; Zeng, F.; Vashisht, A.A.; Wohlschlegel, J.A.; Quintana, D.G. Three Different Pathways Prevent Chromosome Segregation in the Presence of DNA Damage or Replication Stress in Budding Yeast. PLoS Genet. 2015, 11, e1005468. [Google Scholar] [CrossRef] [PubMed]
- Chiker, S.; Pennaneach, V.; Loew, D.; Dingli, F.; Biard, D.; Cordelieres, F.P.; Gemble, S.; Vacher, S.; Bieche, I.; Hall, J.; et al. Cdk5 promotes DNA replication stress checkpoint activation through RPA-32 phosphorylation, and impacts on metastasis free survival in breast cancer patients. Cell Cycle 2015, 14, 3066–3078. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.V.; Dominguez-Sola, D.; Wang, L.C.; Hyrien, O.; Gautier, J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep 2013, 3, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Zanini, I.M.; Herrador, R.; Lopes, M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013, 200, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; Garcia, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.S.; Zhao, R.; Hsu, E.L.; Cayer, J.; Ye, F.; Guo, Y.; Shyr, Y.; Cortez, D. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep. 2010, 11, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Klier, S.; McGowan, C.; Wittenberg, C.; de Bruin, R.A. Chk1 inhibits E2F6 repressor function in response to replication stress to maintain cell-cycle transcription. Curr. Biol. 2013, 23, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Herlihy, A.E.; Pennycook, B.R.; Kriston-Vizi, J.; de Bruin, R.A. Sustained E2F-Dependent Transcription Is a Key Mechanism to Prevent Replication-Stress-Induced DNA Damage. Cell Rep. 2016, 15, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; O’Donohue, M.F.; Wenner, T.; Douarre, C.; Macadre, J.; Koebel, P.; Giraud-Panis, M.J.; Kaplan, H.; Kolkes, A.; Shin-ya, K.; et al. The G-quadruplex ligand telomestatin inhibits POT1 binding to telomeric sequences in vitro and induces GFP-POT1 dissociation from telomeres in human cells. Cancer Res. 2006, 66, 6908–6912. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, T.; Shin-Ya, K.; Sashida, G.; Sumi, M.; Nakajima, A.; Shimamoto, T.; Ohyashiki, J.H.; Ohyashiki, K. Activity of a novel G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), against human leukemia cells: Involvement of ATM-dependent DNA damage response pathways. Oncogene 2003, 22, 5338–5347. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, T.; Shin-ya, K.; Sashida, G.; Sumi, M.; Okabe, S.; Ohyashiki, J.H.; Ohyashiki, K. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: In vitro and in vivo studies in acute leukemia. Oncogene 2006, 25, 5719–5725. [Google Scholar] [CrossRef] [PubMed]
- Temime-Smaali, N.; Guittat, L.; Sidibe, A.; Shin-ya, K.; Trentesaux, C.; Riou, J.F. The G-quadruplex ligand telomestatin impairs binding of topoisomerase IIIalpha to G-quadruplex-forming oligonucleotides and uncaps telomeres in ALT cells. PLoS ONE 2009, 4, e6919. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, D.; Okabe, S.; Okamoto, K.; Nakano, I.; Shin-ya, K.; Seimiya, H. G-quadruplex ligand-induced DNA damage response coupled with telomere dysfunction and replication stress in glioma stem cells. Biochem. Biophys. Res. Commun. 2016, 471, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.M.; Lindsay, H.D. DNA replication stress and cancer: Cause or cure? Future Oncol. 2016, 12, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Puigvert, J.C.; Sanjiv, K.; Helleday, T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 2016, 283, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Jones, R.M.; Higgs, M.R.; Petermann, E. Cancer therapy and replication stress: Forks on the road to perdition. Adv. Clin. Chem. 2015, 69, 91–138. [Google Scholar] [PubMed]
- Rizzo, A.; Salvati, E.; Porru, M.; D’Angelo, C.; Stevens, M.F.; D’Incalci, M.; Leonetti, C.; Gilson, E.; Zupi, G.; Biroccio, A. Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res. 2009, 37, 5353–5364. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Vasquez, K.M. Effects of Replication and Transcription on DNA Structure-Related Genetic Instability. Genes 2017, 8, 17. https://doi.org/10.3390/genes8010017
Wang G, Vasquez KM. Effects of Replication and Transcription on DNA Structure-Related Genetic Instability. Genes. 2017; 8(1):17. https://doi.org/10.3390/genes8010017
Chicago/Turabian StyleWang, Guliang, and Karen M. Vasquez. 2017. "Effects of Replication and Transcription on DNA Structure-Related Genetic Instability" Genes 8, no. 1: 17. https://doi.org/10.3390/genes8010017
APA StyleWang, G., & Vasquez, K. M. (2017). Effects of Replication and Transcription on DNA Structure-Related Genetic Instability. Genes, 8(1), 17. https://doi.org/10.3390/genes8010017