
Mechanisms Governing DDK Regulation of the Initiation of DNA Replication


{kind=link}
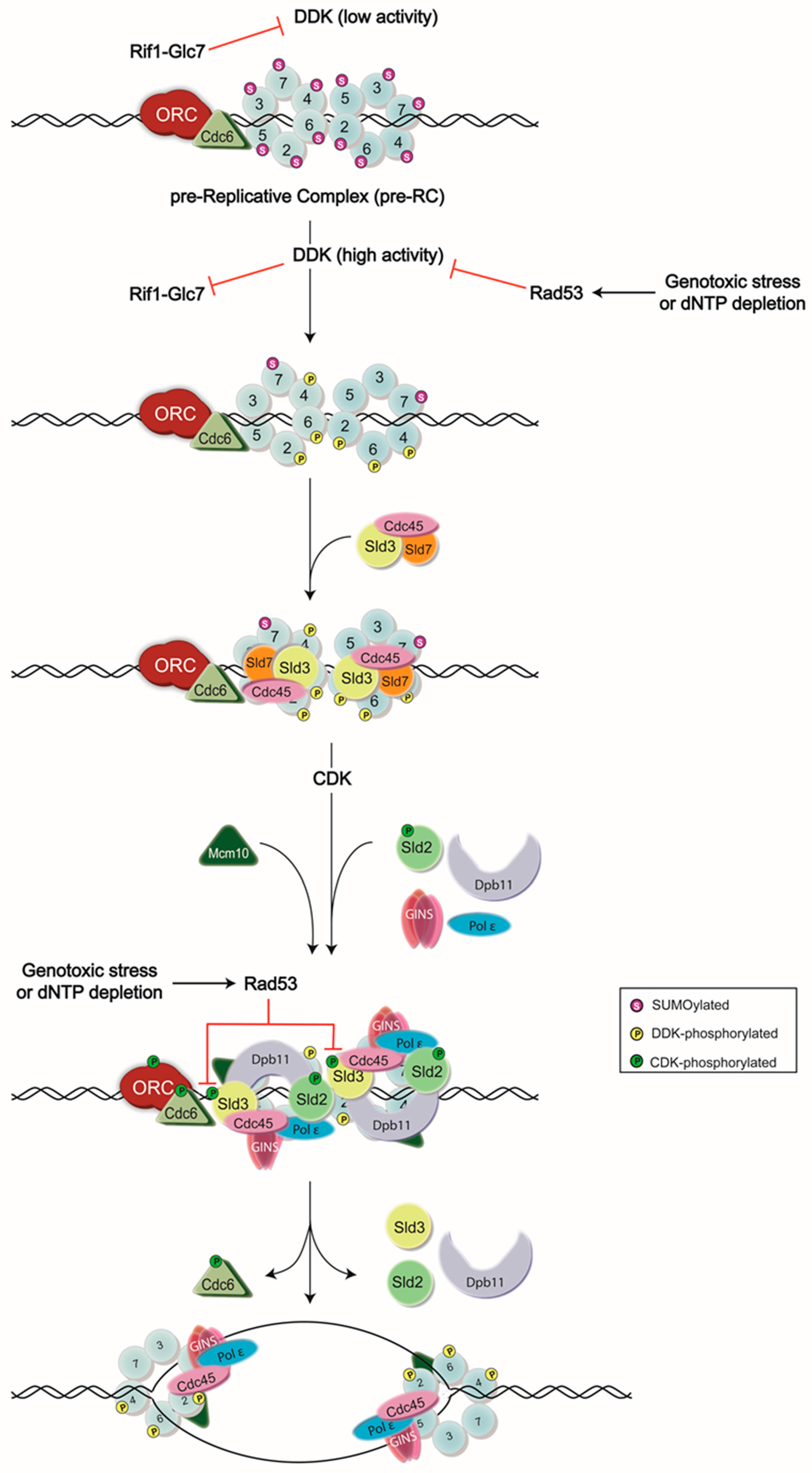
Abstract
:1. Introduction
2. Insights into DDK Interactions with Mcm2-7
3. Regulation of DDK Activity by Rad53
4. Mutual Promotion of Sld3 and DDK Activities
5. Opposing Activities of SUMOylation, Rif1, and DDK
6. Targeting of DDK to Early Replicating Centromeric Origins of DNA Replication
7. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Labib, K. How Do Cdc7 and Cyclin-Dependent Kinases Trigger the Initiation of Chromosome Replication in Eukaryotic Cells? Genes Dev. 2010, 24, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Pasero, P.; Duncker, B.P.; Schwob, E.; Gasser, S.M. A Role for the Cdc7 Kinase Regulatory Subunit Dbf4p in the Formation of Initiation-Competent Origins of Replication. Genes Dev. 1999, 13, 2159–2176. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F.X. Checkpoint-Dependent Inhibition of DNA Replication Initiation by Sld3 and Dbf4 Phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mosqueda, J.; Maas, N.L.; Jonsson, Z.O.; DeFazio-Eli, L.G.; Wohlschlegel, J.; Toczyski, D.P. Damage-Induced Phosphorylation of Sld3 Is Important to Block Late Origin Firing. Nature 2010, 467, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Duch, A.; Palou, G.; Jonsson, Z.O.; Palou, R.; Calvo, E.; Wohlschlegel, J.; Quintana, D.G. A Dbf4 Mutant Contributes to Bypassing the Rad53-Mediated Block of Origins of Replication in Response to Genotoxic Stress. J. Biol. Chem. 2011, 286, 2486–2491. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Kenworthy, J.; Gabrielse, C.; Hänni, C.; Zegerman, P.; Weinreich, M. DNA Replication Checkpoint Signaling Depends on a Rad53–Dbf4 N-Terminal Interaction in Saccharomyces cerevisiae. Genetics 2013, 194, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Kitada, K.; Johnston, L.H.; Sugino, T.; Sugino, A. Temperature-Sensitive cdc7 Mutations of Saccharomyces cerevisiae Are Suppressed by the DBF4 Gene, Which Is Required for the G1/S Cell Cycle Transition. Genetics 1992, 131, 21–29. [Google Scholar] [PubMed]
- Heller, R.C.; Kang, S.; Lam, W.M.; Chen, S.; Chan, C.S.; Bell, S.P. Eukaryotic Origin-Dependent DNA Replication In Vitro Reveals Sequential Action of DDK and S-CDK Kinases. Cell 2011, 146, 80–91. [Google Scholar] [CrossRef] [PubMed]
- On, K.F.; Beuron, F.; Frith, D.; Snijders, A.P.; Morris, E.P.; Diffley, J.F.X. Prereplicative Complexes Assembled In Vitro Support Origin-Dependent and Independent DNA Replication. EMBO J. 2014, 33, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.C.; Tognetti, S.; Riera, A.; Zech, J.; Clarke, P.; Fernández-Cid, A.; Speck, C. A Reconstituted System Reveals How Activating and Inhibitory Interactions Control DDK Dependent Assembly of the Eukaryotic Replicative Helicase. Nucleic Acids Res. 2015, 43, 10238–10250. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.P.; Deegan, T.D.; Janska, A.; Early, A.; Diffley, J.F.X. Regulated Eukaryotic DNA Replication Origin Firing with Purified Proteins. Nature 2015, 519, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Collyer, T.; Hardy, C.F.J. Cell Cycle Regulation of DNA Replication Initiator Factor Dbf4p. Mol. Cell. Biol. 1999, 19, 4270–4278. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, G.; Owens, J.C.; Shellman, Y.; Sclafani, R.A.; Li, J.J. Cell Cycle Control of Cdc7p Kinase Activity through Regulation of Dbf4p Stability. Mol. Cell. Biol. 1999, 19, 4888–4896. [Google Scholar] [CrossRef] [PubMed]
- Weinreich, M.; Stillman, B. Cdc7p-Dbf4p Kinase Binds to Chromatin during S Phase and Is Regulated by Both the APC and the RAD53 Checkpoint Pathway. EMBO J. 1999, 18, 5334–5346. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.F.; Santocanale, C.; Drury, L.S.; Diffley, J.F. Dbf4p, an Essential S Phase-Promoting Factor, Is Targeted for Degradation by the Anaphase-Promoting Complex. Mol. Cell. Biol. 2000, 20, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, S.; Alvino, G.M.; Chang, F.; Lian, H.; Sridhar, A.; Kubota, T.; Brewer, B.J.; Weinreich, M.; Raghuraman, M.K.; Donaldson, A.D. Rif1 Controls DNA Replication by Directing Protein Phosphatase 1 to Reverse Cdc7-Mediated Phosphorylation of the MCM Complex. Genes Dev. 2014, 28, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Mattarocci, S.; Shyian, M.; Lemmens, L.; Damay, P.; Altintas, D.M.; Shi, T.; Bartholomew, C.R.; Thomä, N.H.; Hardy, C.F.J.; Shore, D. Rif1 Controls DNA Replication Timing in Yeast through the PP1 Phosphatase Glc7. Cell Rep. 2014, 7, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.F.; Dryga, O.; Seematter, S.; Pahl, P.M.; Sclafani, R.A. mcm5/cdc46-bob1 Bypasses the Requirement for the S Phase Activator Cdc7p. Proc. Natl. Acad. Sci. USA 1997, 94, 3151–3155. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.L.; Leon, R.P.; Pessoa-Brandao, L.; Hunt, S.; Raghuraman, M.K.; Fangman, W.L.; Brewer, B.J.; Sclafani, R.A. Structural Changes in Mcm5 Protein Bypass Cdc7-Dbf4 Function and Reduce Replication Origin Efficiency in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007, 27, 7594–7602. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.-J.; Stillman, B. Cdc7-Dbf4 Phosphorylates MCM Proteins via a Docking Site-Mediated Mechanism to Promote S Phase Progression. Mol. Cell 2006, 24, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.-J.; Stillman, B. The Dbf4-Cdc7 Kinase Promotes S Phase by Alleviating an Inhibitory Activity in Mcm4. Nature 2010, 463, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.-J.; Kinney, J.B.; Lengronne, A.; Pasero, P.; Stillman, B. Domain within the Helicase Subunit Mcm4 Integrates Multiple Kinase Signals to Control DNA Replication Initiation and Fork Progression. Proc. Natl. Acad. Sci. USA 2014, 111, E1899–E1908. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.-J.; Kinney, J.B.; Stillman, B. Concerted Activities of Mcm4, Sld3, and Dbf4 in Control of Origin Activation and DNA Replication Fork Progression. Genome Res. 2016, 26, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Bruck, I.; Kaplan, D. Dbf4-Cdc7 Phosphorylation of Mcm2 Is Required for Cell Growth. J. Biol. Chem. 2009, 284, 28823–28831. [Google Scholar] [CrossRef] [PubMed]
- Stead, B.E.; Brandl, C.J.; Davey, M.J. Phosphorylation of Mcm2 Modulates Mcm2-7 Activity and Affects the Cell’s Response to DNA Damage. Nucleic Acids Res. 2011, 39, 6998–7008. [Google Scholar] [CrossRef] [PubMed]
- Bruck, I.; Kaplan, D.L. The Dbf4-Cdc7 Kinase Promotes Mcm2-7 Ring Opening to Allow for Single-Stranded DNA Extrusion and Helicase Assembly. J. Biol. Chem. 2015, 290, 1210–1221. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Schwacha, A. The Saccharomyces cerevisiae Mcm6/2 and Mcm5/3 ATPase Active Sites Contribute to the Function of the Putative Mcm2-7 “Gate”. Nucleic Acids Res. 2010, 38, 6078–6088. [Google Scholar] [CrossRef] [PubMed]
- Samel, S.A.; Fernández-Cid, A.; Sun, J.; Riera, A.; Tognetti, S.; Herrera, M.C.; Li, H.; Speck, C. A Unique DNA Entry Gate Serves for Regulated Loading of the Eukaryotic Replicative Helicase MCM2-7 onto DNA. Genes Dev. 2014, 28, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhai, Y.; Zhang, Y.; Li, W.; Yang, M.; Lei, J.; Tye, B.-K.; Gao, N. Structure of the Eukaryotic MCM Complex at 3.8 Å. Nature 2015, 524, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Ilves, I.; Tamberg, N.; Petojevic, T.; Nogales, E.; Botchan, M.R.; Berger, J.M. The Structural Basis for MCM2-7 Helicase Activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 2011, 18, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Kawasaki, Y.; Young, M.R.; Kihara, M.; Sugino, A.; Tye, B.K. Mcm2 Is a Target of Regulation by Cdc7-Dbf4 during the Initiation of DNA Synthesis. Genes Dev. 1997, 11, 3365–3374. [Google Scholar] [CrossRef] [PubMed]
- Randell, J.C.W.; Fan, A.; Chan, C.; Francis, L.I.; Heller, R.C.; Galani, K.; Bell, S.P. Mec1 Is One of Multiple Kinases That Prime the Mcm2-7 Helicase for Phosphorylation by Cdc7. Mol. Cell 2010, 40, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Ramer, M.D.; Suman, E.S.; Richter, H.; Stanger, K.; Spranger, M.; Bieberstein, N.; Duncker, B.P. Dbf4 and Cdc7 Proteins Promote DNA Replication through Interactions with Distinct Mcm2-7 Protein Subunits. J. Biol. Chem. 2013, 288, 14926–14935. [Google Scholar] [CrossRef] [PubMed]
- Francis, L.I.; Randell, J.C.W.; Takara, T.J.; Uchima, L.; Bell, S.P. Incorporation into the Prereplicative Complex Activates the Mcm2-7 Helicase for Cdc7-Dbf4 Phosphorylation. Genes Dev. 2009, 23, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Fernandez-Cid, A.; Riera, A.; Tognetti, S.; Yuan, Z.; Stillman, B.; Speck, C.; Li, H. Structural and Mechanistic Insights into Mcm2-7 Double-Hexamer Assembly and Function. Genes Dev. 2014, 28, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Varrin, A.E.; Prasad, A.A.; Scholz, R.-P.; Ramer, M.D.; Duncker, B.P. A Mutation in Dbf4 Motif M Impairs Interactions with DNA Replication Factors and Confers Increased Resistance to Genotoxic Agents. Mol. Cell. Biol. 2005, 25, 7494–7504. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R.; Prasad, A.A.; Chan, P.K.; Duncker, B.P. The Dbf4 Motif C Zinc Finger Promotes DNA Replication and Mediates Resistance to Genotoxic Stress. Cell Cycle 2010, 9, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.M.; Kawasaki, Y.; Chen, Y.; Lei, M.; Tye, B.K. A Lesion in the DNA Replication Initiation Factor Mcm10 Induces Pausing of Elongation Forks through Chromosomal Replication Origins in Saccharomyces cerevisiae. Mol. Cell. Biol. 1997, 17, 3261–3271. [Google Scholar] [CrossRef] [PubMed]
- Perez-Arnaiz, P.; Bruck, I.; Kaplan, D.L. Mcm10 Coordinates the Timely Assembly and Activation of the Replication Fork Helicase. Nucleic Acids Res. 2016, 44, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-K.; Seo, Y.-S.; Hurwitz, J. The Cdc23 (Mcm10) Protein Is Required for the Phosphorylation of Minichromosome Maintenance Complex by the Dfp1-Hsk1 Kinase. Proc. Natl. Acad. Sci. USA 2003, 100, 2334–2339. [Google Scholar] [CrossRef] [PubMed]
- Homesley, L.; Lei, M.; Kawasaki, Y.; Sawyer, S.; Christensen, T.; Tye, B.K. Mcm10 and the MCM2-7 Complex Interact to Initiate DNA Synthesis and to Release Replication Factors from Origins. Genes Dev. 2000, 14, 913–926. [Google Scholar] [PubMed]
- Izumi, M.; Yanagi, K.; Mizuno, T.; Yokoi, M.; Kawasaki, Y.; Moon, K.-Y.; Hurwitz, J.; Yatagai, F.; Hanaoka, F. The Human Homolog of Saccharomyces cerevisiae Mcm10 Interacts with Replication Factors and Dissociates from Nuclease-Resistant Nuclear Structures in G2 Phase. Nucleic Acids Res. 2000, 28, 4769–4777. [Google Scholar] [CrossRef] [PubMed]
- Liachko, I.; Tye, B.K. Mcm10 Mediates the Interaction between DNA Replication and Silencing Machineries. Genetics 2009, 181, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, F.; Sengupta, S.; De Piccoli, G.; Sanchez-Diaz, A.; Labib, K. Mcm10 Associates with the Loaded DNA Helicase at Replication Origins and Defines a Novel Step in Its Activation. EMBO J. 2012, 31, 2195–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, M.E.; Diffley, J.F.X. Recruitment of Mcm10 to Sites of Replication Initiation Requires Direct Binding to the Minichromosome Maintenance (MCM) Complex. J. Biol. Chem. 2016, 291, 5879–5888. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Xia, Y.; Liu, L.; Cui, J.; Li, Z.; Cao, Q.; Chen, X.S.; Campbell, J.L.; Lou, H. Cell-Cycle-Regulated Interaction between Mcm10 and Double Hexameric Mcm2-7 Is Required for Helicase Splitting and Activation during S Phase. Cell Rep. 2015, 13, 2576–2586. [Google Scholar] [CrossRef] [PubMed]
- Duncker, B.P.; Shimada, K.; Tsai-Pflugfelder, M.; Pasero, P.; Gasser, S.M. An N-Terminal Domain of Dbf4p Mediates Interaction with Both Origin Recognition Complex (ORC) and Rad53p and Can Deregulate Late Origin Firing. Proc. Natl. Acad. Sci. USA 2002, 99, 16087–16092. [Google Scholar] [CrossRef] [PubMed]
- Kihara, M.; Nakai, W.; Asano, S.; Suzuki, A.; Kitada, K.; Kawasaki, Y.; Johnston, L.H.; Sugino, A. Characterization of the Yeast Cdc7p/Dbf4p Complex Purified from Insect Cells. Its Protein Kinase Activity Is Regulated by Rad53p. J. Biol. Chem. 2000, 275, 35051–35062. [Google Scholar] [CrossRef] [PubMed]
- Matthews, L.A.; Jones, D.R.; Prasad, A.A.; Duncker, B.P.; Guarné, A. Saccharomyces cerevisiae Dbf4 Has Unique Fold Necessary for Interaction with Rad53 Kinase. J. Biol. Chem. 2012, 287, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Matthews, L.A.; Selvaratnam, R.; Jones, D.R.; Akimoto, M.; McConkey, B.J.; Melacini, G.; Duncker, B.P.; Guarné, A. A Novel Non-Canonical Forkhead-Associated (FHA) Domain-Binding Interface Mediates the Interaction between Rad53 and Dbf4 Proteins. J. Biol. Chem. 2014, 289, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Aucher, W.; Becker, E.; Ma, E.; Miron, S.; Martel, A.; Ochsenbein, F.; Marsolier-Kergoat, M.-C.; Guerois, R. A Strategy for Interaction Site Prediction between Phospho-Binding Modules and Their Partners Identified from Proteomic Data. Mol. Cell. Proteom. 2010, 9, 2745–2759. [Google Scholar] [CrossRef] [PubMed]
- Almawi, A.W.; Matthews, L.A.; Larasati; Myrox, P.; Boulton, S.; Lai, C.; Moraes, T.; Melacini, G.; Ghirlando, R.; Duncker, B.P.; et al. “AND” Logic Gates at Work: Crystal Structure of Rad53 Bound to Dbf4 and Cdc7. Sci. Rep. 2016, 6, 34237. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Umemori, T.; Hirai, K.; Muramatsu, S.; Kamimura, Y.; Araki, H. CDK-Dependent Phosphorylation of Sld2 and Sld3 Initiates DNA Replication in Budding Yeast. Nature 2007, 445, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F.X. Phosphorylation of Sld2 and Sld3 by Cyclin-Dependent Kinases Promotes DNA Replication in Budding Yeast. Nature 2007, 445, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nakato, R.; Katou, Y.; Shirahige, K.; Araki, H. Origin Association of Sld3, Sld7, and Cdc45 Proteins Is a Key Step for Determination of Origin-Firing Timing. Curr. Biol. 2011, 21, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, Y.; Tak, Y.-S.; Sugino, A.; Araki, H. Sld3, Which Interacts with Cdc45 (Sld4), Functions for Chromosomal DNA Replication in Saccharomyces cerevisiae. EMBO J. 2001, 20, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Mantiero, D.; Mackenzie, A.; Donaldson, A.; Zegerman, P. Limiting Replication Initiation Factors Execute the Temporal Programme of Origin Firing in Budding Yeast. EMBO J. 2011, 30, 4805–4814. [Google Scholar] [CrossRef] [PubMed]
- Bruck, I.; Kaplan, D.L. GINS and Sld3 Compete with One Another for Mcm2-7 and Cdc45 Binding. J. Biol. Chem. 2011, 286, 14157–14167. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Umemori, T.; Endo, S.; Muramatsu, S.; Kanemaki, M.; Kamimura, Y.; Obuse, C.; Araki, H. Sld7, an Sld3-associated Protein Required for Efficient Chromosomal DNA Replication in Budding Yeast. EMBO J. 2011, 30, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, H.; Yamada, Y.; Uchida, T.; Sunathvanichkul, T.; Nakagawa, T.; Masukata, H. Ordered Assembly of Sld3, GINS and Cdc45 Is Distinctly Regulated by DDK and CDK for Activation of Replication Origins. EMBO J. 2006, 25, 4663–4674. [Google Scholar] [CrossRef] [PubMed]
- Deegan, T.D.; Yeeles, J.T.; Diffley, J.F. Phosphopeptide Binding by Sld3 Links Dbf4-Dependent Kinase to MCM Replicative Helicase Activation. EMBO J. 2016, 35, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Cao, Q.; Lou, H. Sld3-MCM Interaction Facilitated by Dbf4-Dependent Kinase Defines an Essential Step in Eukaryotic DNA Replication Initiation. Front. Microbiol. 2016, 7, 885. [Google Scholar] [CrossRef] [PubMed]
- Itou, H.; Muramatsu, S.; Shirakihara, Y.; Araki, H. Crystal Structure of the Homology Domain of the Eukaryotic DNA Replication Proteins Sld3/Treslin. Structure 2014, 22, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Bruck, I.; Kaplan, D.L. Conserved Mechanism for Coordinating Replication Fork Helicase Assembly with Phosphorylation of the Helicase. Proc. Natl. Acad. Sci. USA 2015, 112, 11223–11228. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.F.; Sussel, L.; Shore, D. A RAP1-Interacting Protein Involved in Transcriptional Silencing and Telomere Length Regulation. Genes Dev. 1992, 6, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.-Y.; Robertson, E.D.; Hiraga, S.; Alvino, G.M.; Collingwood, D.; McCune, H.J.; Sridhar, A.; Brewer, B.J.; Raghuraman, M.K.; Donaldson, A.D. The Effect of Ku on Telomere Replication Time Is Mediated by Telomere Length but Is Independent of Histone Tail Acetylation. Mol. Biol. Cell 2011, 22, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Cornacchia, D.; Dileep, V.; Quivy, J.-P.; Foti, R.; Tili, F.; Santarella-Mellwig, R.; Antony, C.; Almouzni, G.; Gilbert, D.M.; Buonomo, S.B.C. Mouse Rif1 Is a Key Regulator of the Replication-Timing Programme in Mammalian Cells. EMBO J. 2012, 31, 3678–3690. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Kanoh, Y.; Matsumoto, S.; Renard-Guillet, C.; Shirahige, K.; Masai, H. Rif1 Is a Global Regulator of Timing of Replication Origin Firing in Fission Yeast. Genes Dev. 2012, 26, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 Regulates the Replication Timing Domains on the Human Genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Davé, A.; Cooley, C.; Garg, M.; Bianchi, A. Protein Phosphatase 1 Recruitment by Rif1 Regulates DNA Replication Origin Firing by Counteracting DDK Activity. Cell Rep. 2014, 7, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhao, X. A New MCM Modification Cycle Regulates DNA Replication Initiation. Nat. Struct. Mol. Biol. 2016, 23, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, M.K.; Winzeler, E.A.; Collingwood, D.; Hunt, S.; Wodicka, L.; Conway, A.; Lockhart, D.J.; Davis, R.W.; Brewer, B.J.; Fangman, W.L. Replication Dynamics of the Yeast Genome. Science 2001, 294, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Dubey, D.D.; Huberman, J.A. Early-Replicating Heterochromatin. Genes Dev. 2003, 17, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Tsai, H.-J.; Tirosh, I.; Burrack, L.S.; Barkai, N.; Berman, J. Epigenetically-Inherited Centromere and Neocentromere DNA Replicates Earliest in S-Phase. PLoS Genet. 2010, 6, e1001068. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.A.; Nieduszynski, C.A. Conservation of Replication Timing Reveals Global and Local Regulation of Replication Origin Activity. Genome Res. 2012, 22, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Tiengwe, C.; Marcello, L.; Farr, H.; Dickens, N.; Kelly, S.; Swiderski, M.; Vaughan, D.; Gull, K.; Barry, J.D.; Bell, S.D.; et al. Genome-Wide Analysis Reveals Extensive Functional Interaction between DNA Replication Initiation and Transcription in the Genome of Trypanosoma brucei. Cell Rep. 2012, 2, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Henikoff, S. Centromeres Are Specialized Replication Domains in Heterochromatin. J. Cell Biol. 2001, 153, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Natsume, T.; Müller, C.A.; Katou, Y.; Retkute, R.; Gierliński, M.; Araki, H.; Blow, J.J.; Shirahige, K.; Nieduszynski, C.A.; Tanaka, T.U. Kinetochores Coordinate Pericentromeric Cohesion and Early DNA Replication by Cdc7-Dbf4 Kinase Recruitment. Mol. Cell 2013, 50, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.S.; Basu, A.; Bermudez, V.; Hurwitz, J.; Walter, J.C. Cdc7-Drf1 Kinase Links Chromosome Cohesion to the Initiation of DNA Replication in Xenopus Egg Extracts. Genes Dev. 2008, 22, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.Z.L.; Wang, G.-N.; Fitzgerald, J.; Quachthithu, H.; Rainey, M.D.; Cattaneo, A.; Bachi, A.; Santocanale, C. DDK Dependent Regulation of TOP2A at Centromeres Revealed by a Chemical Genetics Approach. Nucleic Acids Res. 2016, 44, 8786–8798. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larasati; Duncker, B.P. Mechanisms Governing DDK Regulation of the Initiation of DNA Replication. Genes 2017, 8, 3. https://doi.org/10.3390/genes8010003
Larasati, Duncker BP. Mechanisms Governing DDK Regulation of the Initiation of DNA Replication. Genes. 2017; 8(1):3. https://doi.org/10.3390/genes8010003
Chicago/Turabian StyleLarasati, and Bernard P. Duncker. 2017. "Mechanisms Governing DDK Regulation of the Initiation of DNA Replication" Genes 8, no. 1: 3. https://doi.org/10.3390/genes8010003
APA StyleLarasati, & Duncker, B. P. (2017). Mechanisms Governing DDK Regulation of the Initiation of DNA Replication. Genes, 8(1), 3. https://doi.org/10.3390/genes8010003