
RNA Editing, ADAR1, and the Innate Immune Response

{kind=link}
{kind=link}
Abstract
:1. ADAR1 and RNA Editing
2. ADAR1 and IFN Signaling
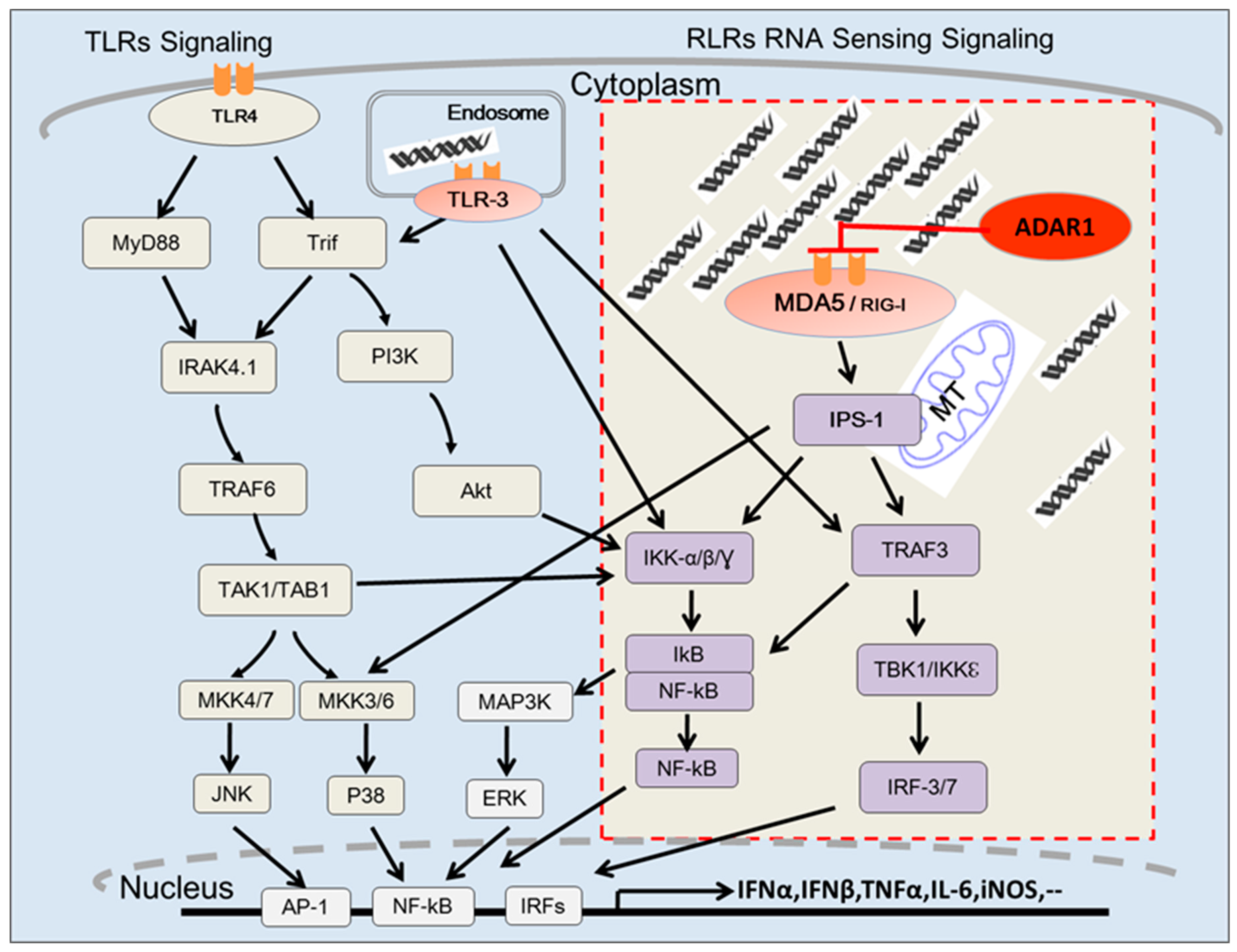
3. ADAR1 Suppresses the Sensing of Endogenous RNA by Cytosolic dsRNA Receptors
4. RNA Editing Activity of ADAR1 and the Innate Immune Response
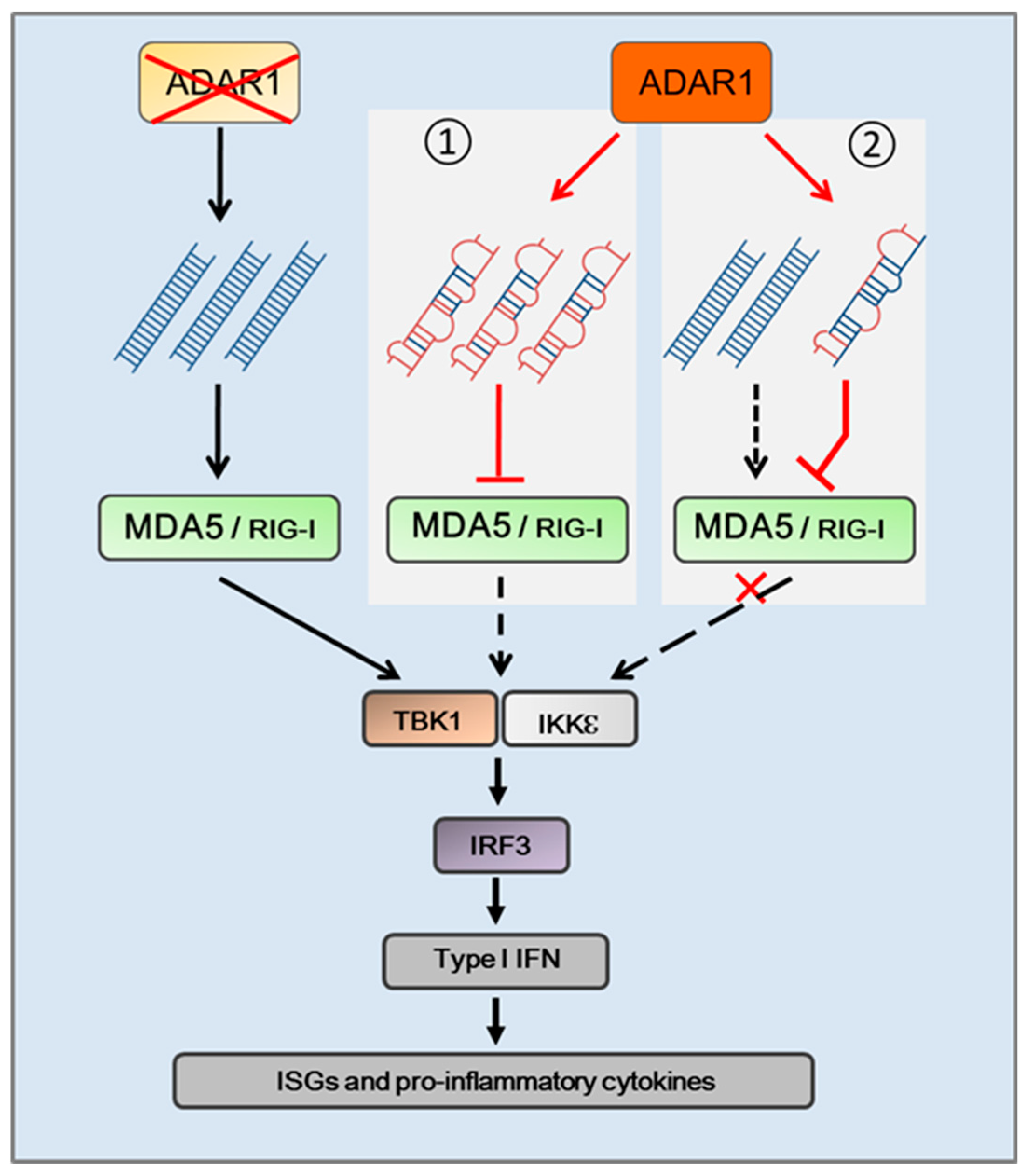
5. Cytosolic RNA Receptors Responding to Cellular “Self” RNA of ADAR1 Substrate
6. ADAR1 Edited Cellular dsRNA and Innate Immune Response
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hough, R.F.; Bass, B.L. Purification of the xenopus laevis double-stranded RNA adenosine deaminase. J. Biol. Chem. 1994, 269, 9933–9939. [Google Scholar] [PubMed]
- Bass, B.L.; Weintraub, H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef]
- Kim, U.; Garner, T.L.; Sanford, T.; Speicher, D.; Murray, J.M.; Nishikura, K. Purification and characterization of double-stranded RNA adenosine deaminase from bovine nuclear extracts. J. Biol. Chem. 1994, 269, 13480–13489. [Google Scholar] [PubMed]
- Kim, U.; Wang, Y.; Sanford, T.; Zeng, Y.; Nishikura, K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. USA 1994, 91, 11457–11461. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.B.; Samuel, C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol. Cell. Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; Samuel, C.E. Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene 1999, 229, 203–213. [Google Scholar] [CrossRef]
- George, C.X.; Samuel, C.E. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl. Acad. Sci. USA 1999, 96, 4621–4626. [Google Scholar] [CrossRef] [PubMed]
- Melcher, T.; Maas, S.; Herb, A.; Sprengel, R.; Seeburg, P.H.; Higuchi, M. A mammalian RNA editing enzyme. Nature 1996, 379, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Kohr, G.; Melcher, T.; Seeburg, P.H. Candidate editases for glur channels in single neurons of rat hippocampus and cerebellum. Neuropharmacology 1998, 37, 1411–1417. [Google Scholar] [CrossRef]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Melcher, T.; Maas, S.; Herb, A.; Sprengel, R.; Higuchi, M.; Seeburg, P.H. Red2, a brain-specific member of the RNA-specific adenosine deaminase family. J. Biol. Chem. 1996, 271, 31795–31798. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and regulation of RNA editing by adar deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, P.H. A-to-I editing: New and old sites, functions and speculations. Neuron 2002, 35, 17–20. [Google Scholar] [CrossRef]
- Knight, S.W.; Bass, B.L. The role of RNA editing by ADARS in RNAi. Mol. Cell. 2002, 10, 809–817. [Google Scholar] [CrossRef]
- Nishikura, K. Editor meets silencer: Crosstalk between RNA editing and RNA interference. Nat. Rev. Mol. Cell. Biol. 2006, 7, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Gott, J.M.; Emeson, R.B. Functions and mechanisms of RNA editing. Annu. Rev. Genet. 2000, 34, 499–531. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, P.H.; Hartner, J. Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr. Opin. Neurobiol. 2003, 13, 279–283. [Google Scholar] [CrossRef]
- Mallela, A.; Nishikura, K. A-to-I editing of protein coding and noncoding RNAs. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Hogg, M.; Paro, S.; Keegan, L.P.; O′Connell, M.A. RNA editing by mammalian ADARS. Adv. Genet. 2011, 73, 87–120. [Google Scholar] [PubMed]
- Ohlson, J.; Pedersen, J.S.; Haussler, D.; Ohman, M. Editing modifies the GABA(A) receptor subunit alpha3. RNA 2007, 13, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, T.; Rosenthal, J.J.; Holmgren, M.; Reenan, R. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat. Struct. Mol. Biol. 2004, 11, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.M.; Chu, H.; Rueter, S.M.; Hutchinson, L.K.; Canton, H.; Sanders-Bush, E.; Emeson, R.B. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 1997, 387, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an ampa receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wang, Q.; Howell, K.L.; Lee, J.T.; Cho, D.S.; Murray, J.M.; Nishikura, K. ADAR1 RNA deaminase limits short interfering RNA efficacy in mammalian cells. J. Biol. Chem. 2005, 280, 3946–3953. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, J.J.; Seeburg, P.H. A-to-I RNA editing: Effects on proteins key to neural excitability. Neuron 2012, 74, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; Emeson, R.B. Editing of neurotransmitter receptor and ion channel RNAs in the nervous system. Curr. Top. Microbiol. Immunol. 2012, 353, 61–90. [Google Scholar] [PubMed]
- Porath, H.T.; Carmi, S.; Levanon, E.Y. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of alu-containing mrnas in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blow, M.; Futreal, P.A.; Wooster, R.; Stratton, M.R. A survey of RNA editing in human brain. Genome Res. 2004, 14, 2379–2387. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.D.; Kim, T.T.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Lagergren, J.; Ohman, M. RNA editing of non-coding RNA and its role in gene regulation. Biochimie 2015, 117, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing let-7 biogenesis. Cell Stem Cell. 2016, 19, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, E.; Mobley, A.K.; Braeuer, R.R.; Kamiya, T.; Huang, L.; Vasquez, M.E.; Salameh, A.; Lee, H.J.; Kim, S.J.; Ivan, C.; et al. Reduced adenosine-to-inosine mir-455–5p editing promotes melanoma growth and metastasis. Nat. Cell. Biol. 2015, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hui, H.; Guo, Z.; Zhang, W.; Hu, Y.; He, T.; Tai, Y.; Peng, P.; Wang, L. ADAR1 regulates arhgap26 gene expression through RNA editing by disrupting mir-30b-3p and mir-573 binding. RNA 2013, 19, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Stellos, K.; Gatsiou, A.; Stamatelopoulos, K.; Perisic Matic, L.; John, D.; Lunella, F.F.; Jae, N.; Rossbach, O.; Amrhein, C.; Sigala, F.; et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling hur-mediated post-transcriptional regulation. Nat. Med. 2016, 22, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Cui, X.B.; Wang, J.N.; Dong, K.; Chen, S.Y. ADAR1-mediated RNA editing, a novel mechanism controlling phenotypic modulation of vascular smooth muscle cells. Circ. Res. 2016, 119, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Dabiri, G.A.; Lai, F.; Drakas, R.A.; Nishikura, K. Editing of the glur-b ion channel RNA in vitro by recombinant double-stranded rna adenosine deaminase. EMBO J. 1996, 15, 34–45. [Google Scholar] [PubMed]
- Liu, Y.; Samuel, C.E. Editing of glutamate receptor subunit b pre-mrna by splice-site variants of interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J. Biol. Chem. 1999, 274, 5070–5077. [Google Scholar] [CrossRef] [PubMed]
- Wisden, W.; Seeburg, P.H. Mammalian ionotropic glutamate receptors. Curr. Opin. Neurobiol. 1993, 3, 291–298. [Google Scholar] [CrossRef]
- Higuchi, M.; Single, F.N.; Kohler, M.; Sommer, B.; Sprengel, R.; Seeburg, P.H. RNA editing of ampa receptor subunit glur-b: A base-paired intron-exon structure determines position and efficiency. Cell 1993, 75, 1361–1370. [Google Scholar] [CrossRef]
- Sommer, B.; Kohler, M.; Sprengel, R.; Seeburg, P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef]
- Wang, Q.; Khillan, J.; Gadue, P.; Nishikura, K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 2000, 290, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Miyakoda, M.; Yang, W.; Khillan, J.; Stachura, D.L.; Weiss, M.J.; Nishikura, K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J. Biol. Chem. 2004, 279, 4952–4961. [Google Scholar] [CrossRef] [PubMed]
- Hartner, J.C.; Schmittwolf, C.; Kispert, A.; Muller, A.M.; Higuchi, M.; Seeburg, P.H. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 2004, 279, 4894–4902. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Megraw, M.; Kreider, E.; Iizasa, H.; Valente, L.; Hatzigeorgiou, A.G.; Nishikura, K. Frequency and fate of microrna editing in human brain. Nucleic Acids Res. 2008, 36, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.V.; George, C.X.; Welch, M.J.; Liou, L.Y.; Hahm, B.; Lewicki, H.; de la Torre, J.C.; Samuel, C.E.; Oldstone, M.B. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Heale, B.S.; Keegan, L.P.; McGurk, L.; Michlewski, G.; Brindle, J.; Stanton, C.M.; Caceres, J.F.; O′Connell, M.A. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009, 28, 3145–3156. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Ding, L.; Kao, P.N.; Braun, R.; Yang, J.H. ADAR1 interacts with nf90 through double-stranded RNA and regulates nf90-mediated gene expression independently of RNA editing. Mol. Cell. Biol. 2005, 25, 6956–6963. [Google Scholar] [CrossRef] [PubMed]
- Scadden, A.D.; O′Connell, M.A. Cleavage of dsRNAs hyper-edited by adars occurs at preferred editing sites. Nucleic Acids Res. 2005, 33, 5954–5964. [Google Scholar] [CrossRef] [PubMed]
- Valente, L.; Nishikura, K. Adar gene family and A-to-I RNA editing: Diverse roles in posttranscriptional gene regulation. Prog. Nucleic Acid Res. Mol. Biol. 2005, 79, 299–338. [Google Scholar] [PubMed]
- Hundley, H.A.; Bass, B.L. ADAR editing in double-stranded utrs and other noncoding RNA sequences. Trends Biochem. Sci. 2010, 35, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2009, 10, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause aicardi-goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Aicardi, J.; Goutieres, F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann. Neurol. 1984, 15, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Forte, G.M.; Szynkiewicz, M.; Chase, D.S.; Aeby, A.; Abdel-Hamid, M.S.; Ackroyd, S.; Allcock, R.; Bailey, K.M.; Balottin, U.; et al. Assessment of interferon-related biomarkers in aicardi-goutieres syndrome associated with mutations in trex1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet Neurol. 2013, 12, 1159–1169. [Google Scholar] [CrossRef]
- Bonnemann, C.G.; Meinecke, P. Encephalopathy of infancy with intracerebral calcification and chronic spinal fluid lymphocytosis—Another case of the aicardi-goutieres syndrome. Neuropediatrics 1992, 23, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Lebon, P.; Badoual, J.; Ponsot, G.; Goutieres, F.; Hemeury-Cukier, F.; Aicardi, J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J. Neurol. Sci. 1988, 84, 201–208. [Google Scholar] [CrossRef]
- Crow, Y.J.; Hayward, B.E.; Parmar, R.; Robins, P.; Leitch, A.; Ali, M.; Black, D.N.; van Bokhoven, H.; Brunner, H.G.; Hamel, B.C.; et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease trex1 cause aicardi-goutieres syndrome at the ags1 locus. Nat. Genet. 2006, 38, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Leitch, A.; Hayward, B.E.; Garner, A.; Parmar, R.; Griffith, E.; Ali, M.; Semple, C.; Aicardi, J.; Babul-Hirji, R.; et al. Mutations in genes encoding ribonuclease h2 subunits cause aicardi-goutieres syndrome and mimic congenital viral brain infection. Nat. Genet. 2006, 38, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; del Toro Duany, Y.; Jenkinson, E.M.; Forte, G.M.; Anderson, B.H.; Ariaudo, G.; Bader-Meunier, B.; Baildam, E.M.; Battini, R.; Beresford, M.W.; et al. Gain-of-function mutations in ifih1 cause a spectrum of human disease phenotypes associated with upregulated type i interferon signaling. Nat. Genet. 2014, 46, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, B.L. RNA editing. An I for editing. Curr. Biol. 1995, 5, 598–600. [Google Scholar] [CrossRef]
- Bass, B.L. Double-stranded RNA binding proteins and their substrates. Nucleic Acids Symp. Ser. 1995, 13–15. [Google Scholar]
- Yang, S.; Deng, P.; Zhu, Z.; Zhu, J.; Wang, G.; Zhang, L.; Chen, A.F.; Wang, T.; Sarkar, S.N.; Billiar, T.R.; et al. Adenosine deaminase acting on RNA 1 limits rig-I RNA detection and suppresses ifn production responding to viral and endogenous rnas. J. Immunol. 2014, 193, 3436–3445. [Google Scholar] [CrossRef] [PubMed]
- Neeman, Y.; Levanon, E.Y.; Jantsch, M.F.; Eisenberg, E. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 2006, 12, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.B.; Thomis, D.C.; Hans, S.L.; Samuel, C.E. Mechanism of interferon action: Double-stranded RNA-specific adenosine deaminase from human cells is inducible by alpha and gamma interferons. Virology 1995, 210, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.A.; Yang, Q.; Gasparetto, M.; Robinson, L.J.; Liu, X.; Lenzner, D.E.; Hou, J.; Smith, C.; Wang, Q. Deletion of the RNA-editing enzyme ADAR1 causes regression of established chronic myelogenous leukemia in mice. Int. J. Cancer 2013, 132, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Sharma, R.; Nie, D.; Jiao, H.; Im, H.J.; Lai, Y.; Zhao, Z.; Zhu, K.; Fan, J.; Chen, D.; et al. ADAR1 ablation decreases bone mass by impairing osteoblast function in mice. Gene 2013, 513, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Gowen, B.B.; Wong, M.H.; Jung, K.H.; Sanders, A.B.; Mitchell, W.M.; Alexopoulou, L.; Flavell, R.A.; Sidwell, R.W. Tlr3 is essential for the induction of protective immunity against punta toro virus infection by the double-stranded RNA (dsRNA), poly(I:C12u), but not poly(I:C): Differential recognition of synthetic dsrna molecules. J. Immunol. 2007, 178, 5200–5208. [Google Scholar] [CrossRef] [PubMed]
- Cavassani, K.A.; Ishii, M.; Wen, H.; Schaller, M.A.; Lincoln, P.M.; Lukacs, N.W.; Hogaboam, C.M.; Kunkel, S.L. Tlr3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med. 2008, 205, 2609–2621. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of nf-kappab by toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Sousa, C.R.E. Innate recognition of viruses. Immunity 2007, 27, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the ifn-inducible RNA helicase, mda-5, and inhibit its activation of the ifn-beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of rig-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Pestal, K.; Funk, C.C.; Snyder, J.M.; Price, N.D.; Treuting, P.M.; Stetson, D.B. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 2015, 43, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Mannion, N.M.; Greenwood, S.M.; Young, R.; Cox, S.; Brindle, J.; Read, D.; Nellaker, C.; Vesely, C.; Ponting, C.P.; McLaughlin, P.J.; et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell. Rep. 2014, 9, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents mda5 sensing of endogenous dsrna as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, S.; Chen, Z.J. Snapshot: Pathways of antiviral innate immunity. Cell 2010, 140, 436–436.e2. [Google Scholar] [CrossRef] [PubMed]
- Konerman, M.A.; Lok, A.S. Interferon treatment for hepatitis B. Clin. Liver Dis. 2016, 20, 645–665. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, T.L.; Mutimer, D. Hepatitis B: Encouraging the use of interferon. Curr. Opin. Infect. Dis. 2015, 28, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Zamarin, D.; Wolchok, J.D. Immunotherapy of melanoma. Prog. Tumor Res. 2015, 42, 22–29. [Google Scholar] [PubMed]
- Meraz, M.A.; White, J.M.; Sheehan, K.C.; Bach, E.A.; Rodig, S.J.; Dighe, A.S.; Kaplan, D.H.; Riley, J.K.; Greenlund, A.C.; Campbell, D.; et al. Targeted disruption of the stat1 gene in mice reveals unexpected physiologic specificity in the jak-stat signaling pathway. Cell 1996, 84, 431–442. [Google Scholar] [CrossRef]
- Painter, M.M.; Morrison, J.H.; Zoecklein, L.J.; Rinkoski, T.A.; Watzlawik, J.O.; Papke, L.M.; Warrington, A.E.; Bieber, A.J.; Matchett, W.E.; Turkowski, K.L.; et al. Antiviral protection via RDrP-mediated stable activation of innate immunity. PLoS Pathog. 2015, 11, e1005311. [Google Scholar] [CrossRef] [PubMed]
- Vitali, P.; Scadden, A.D. Double-stranded rnas containing multiple iu pairs are sufficient to suppress interferon induction and apoptosis. Nat. Struct. Mol. Biol. 2010, 17, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, T.; Cao, X. RNA editing by ADAR1 marks dsrna as "self". Cell. Res. 2015, 25, 1283–1284. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Chalk, A.M.; Walkley, C.R. Adar1, inosine and the immune sensing system: Distinguishing self from non-self. Wiley Interdiscip. Rev. RNA 2016, 7, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, H.; Singh, S.; Zhou, P.; Yang, S.; Wang, Y.; Zhu, Z.; Zhang, J.; Chen, A.; Billiar, T.; et al. ADAR1 prevents liver injury from inflammation and suppresses interferon production in hepatocytes. Am. J. Pathol. 2015, 185, 3224–3237. [Google Scholar] [PubMed]
- Wang, H.; Wang, G.; Zhang, L.; Zhang, J.; Wang, Q.; Billiar, T.R. ADAR1 suppresses the activation of cytosolic RNA-sensing signaling pathways to protect the liver from ischemia/reperfusion injury. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hundley, H.A.; Krauchuk, A.A.; Bass, B.L. C. Elegans and H. Sapiens mRNAs with edited 3′ UTRs are present on polysomes. RNA 2008, 14, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.S.; Bass, B.L. Inosine exists in mrna at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 1998, 17, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Yano, T.; Kawabata, H.; Ueda, H.; Suzuki, T. Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat. Chem. Biol. 2010, 6, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Li, J.B.; Levanon, E.Y.; Yoon, J.K.; Aach, J.; Xie, B.; Leproust, E.; Zhang, K.; Gao, Y.; Church, G.M. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 2009, 324, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of rna-seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Carmi, S.; Borukhov, I.; Levanon, E.Y. Identification of widespread ultra-edited human RNAs. PLoS Genet. 2011, 7, e1002317. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. Alu elements: Know the sines. Genome Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Blango, M.G.; Bass, B.L. Identification of the long, edited dsRNAome of LPS-stimulated immune cells. Genome Res. 2016, 26, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Harjanto, D.; Papamarkou, T.; Oates, C.J.; Rayon-Estrada, V.; Papavasiliou, F.N.; Papavasiliou, A. RNA editing generates cellular subsets with diverse sequence within populations. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q. RNA editing catalyzed by ADAR1 and its function in mammalian cells. Biochemistry 2011, 76, 900–911. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Li, X.; Qi, R.; Billiar, T. RNA Editing, ADAR1, and the Innate Immune Response. Genes 2017, 8, 41. https://doi.org/10.3390/genes8010041
Wang Q, Li X, Qi R, Billiar T. RNA Editing, ADAR1, and the Innate Immune Response. Genes. 2017; 8(1):41. https://doi.org/10.3390/genes8010041
Chicago/Turabian StyleWang, Qingde, Xiaoni Li, Ruofan Qi, and Timothy Billiar. 2017. "RNA Editing, ADAR1, and the Innate Immune Response" Genes 8, no. 1: 41. https://doi.org/10.3390/genes8010041
APA StyleWang, Q., Li, X., Qi, R., & Billiar, T. (2017). RNA Editing, ADAR1, and the Innate Immune Response. Genes, 8(1), 41. https://doi.org/10.3390/genes8010041