Clinical and Genetic Evaluation of a Cohort of Pediatric Patients with Severe Inherited Retinal Dystrophies



Abstract
:1. Introduction
2. Materials and Methods
2.1. Ophthalmological Analysis
2.2. Clinical Diagnosis
2.3. Selection of RETplex Genes and Enrichment Procedures
2.4. Targeted NGS Analysis
3. Results
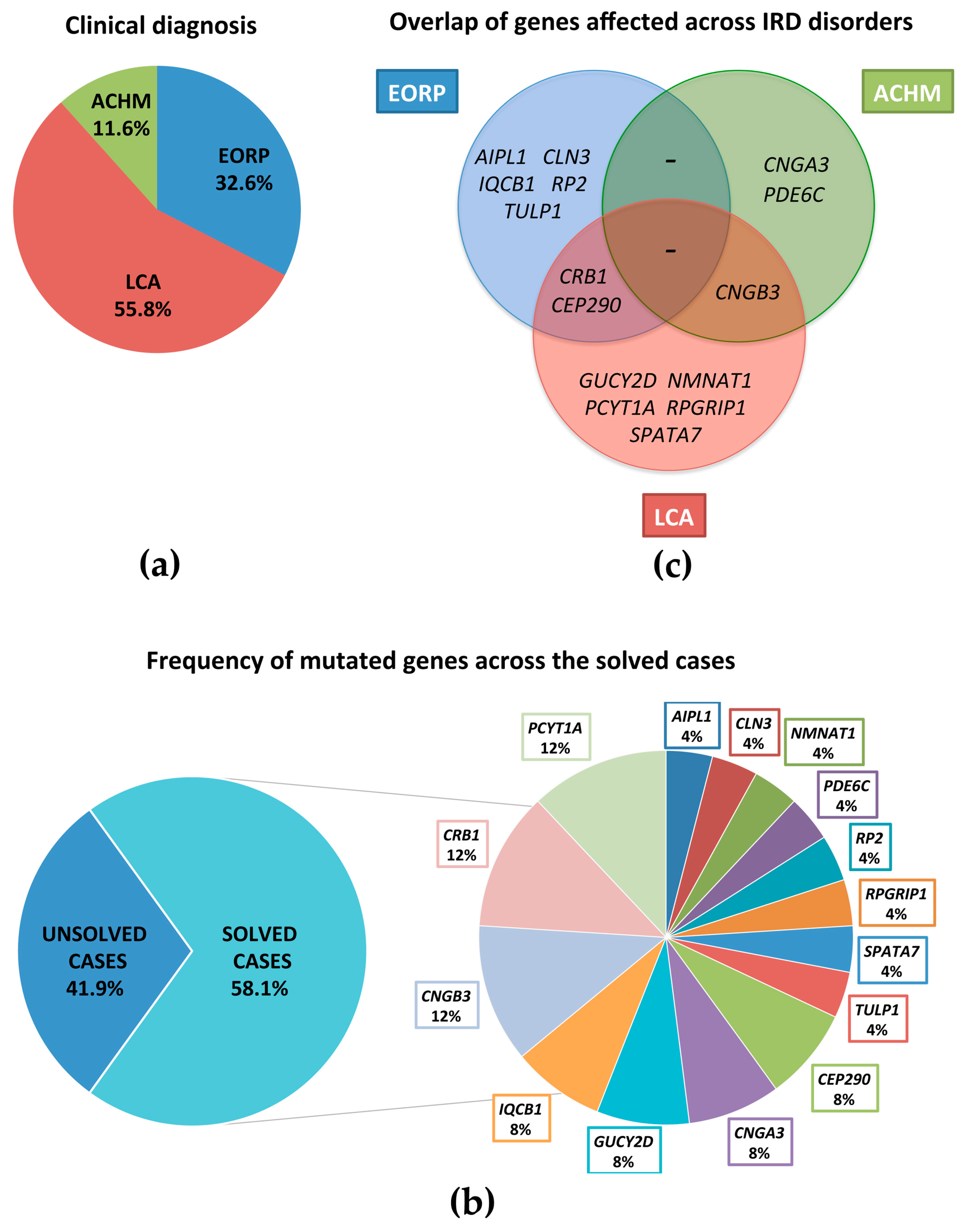
3.1. Patient Selection
3.2. RETplex Analysis
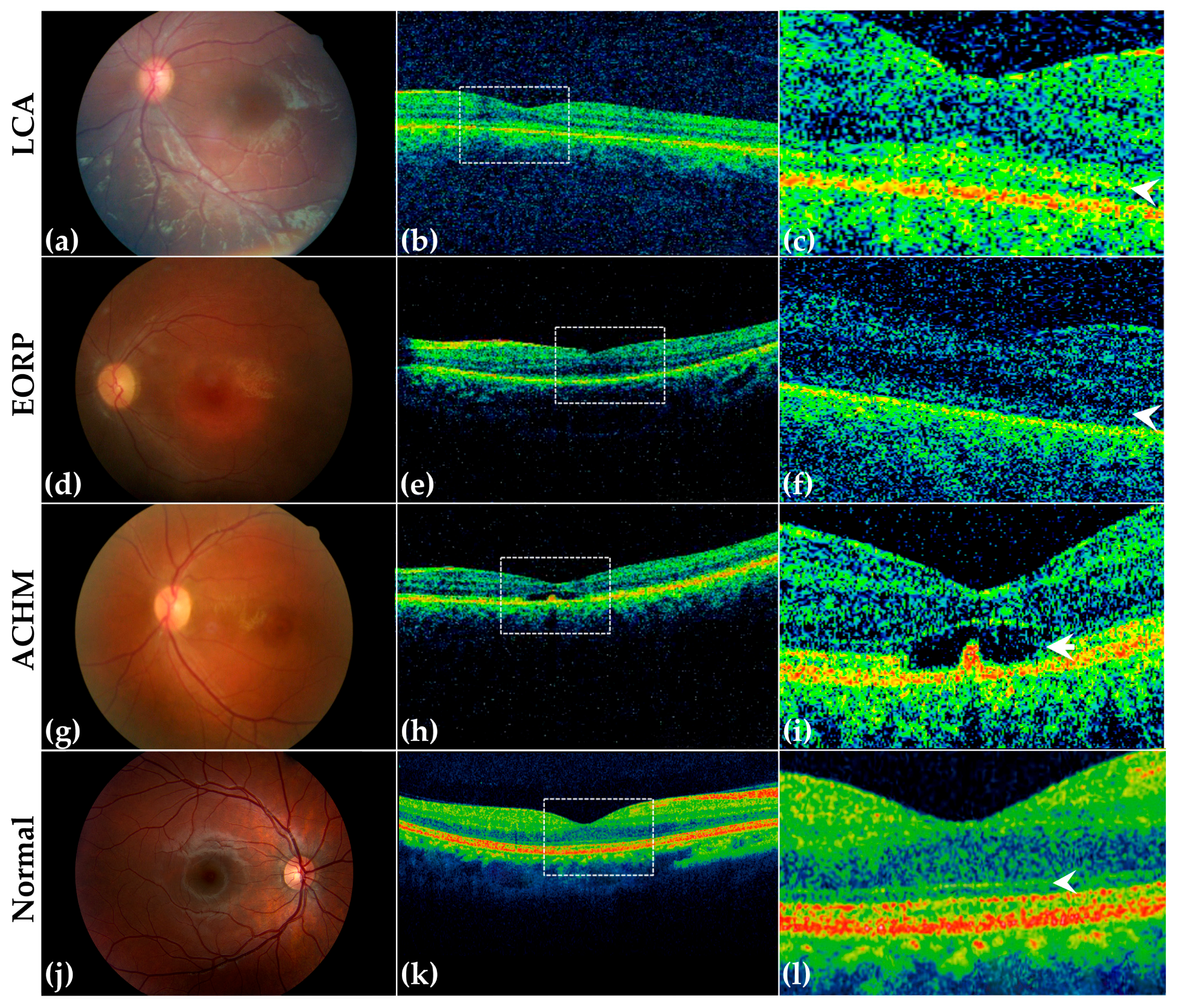
3.3. Clinical Examination
3.3.1. LCA Patients
3.3.2. EORP Patients
3.3.3. ACHM Patients
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, X.F.; Huang, F.; Wu, K.C.; Wu, J.; Chen, J.; Pang, C.P.; Lu, F.; Qu, J.; Jin, Z.B. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Zuchner, S.; Dallman, J.; Wen, R.; Beecham, G.; Naj, A.; Farooq, A.; Kohli, M.A.; Whitehead, P.L.; Hulme, W.; Konidari, I.; et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am. J. Hum. Genet. 2011, 88, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Bujakowska, K.M.; Leveillard, T.; Mohand-Said, S.; Lancelot, M.E.; Germain, A.; Antonio, A.; Michiels, C.; Saraiva, J.P.; Letexier, M.; et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J. Rare Dis. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Shanks, M.E.; Downes, S.M.; Copley, R.R.; Lise, S.; Broxholme, J.; Hudspith, K.A.; Kwasniewska, A.; Davies, W.I.; Hankins, M.W.; Packham, E.R.; et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur. J. Hum. Genet. 2013, 21, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Charbel Issa, P.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLoS ONE 2013, 8, e78496. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Wellman, J.; Marshall, K.A.; McCague, S.; Ashtari, M.; DiStefano-Pappas, J.; Elci, O.U.; Chung, D.C.; Sun, J.; Wright, J.F.; et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: A follow-on phase 1 trial. Lancet 2016, 388, 661–672. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Ratnakaram, R.; Heon, E.; Schwartz, S.B.; Roman, A.J.; Peden, M.C.; Aleman, T.S.; Boye, S.L.; Sumaroka, A.; et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: Safety and efficacy in 15 children and adults followed up to 3 years. Arch. Ophthalmol. 2012, 130, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N., Jr.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.W.; Wu, Z.; Guymer, R.H.; Luu, C.D. Ellipsoid zone on optical coherence tomography: A review. Clin Exp. Ophthalmol. 2016, 44, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. Adv. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Camacho, O.F.; Zenteno, J.C. Review and update on the molecular basis of Leber congenital amaurosis. World J. Clin. Cases 2015, 3, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Jagle, H.; Wissinger, B. Achromatopsia. In GeneReviews®; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.R.; Parfitt, D.A.; Gardner, J.C.; Martinez, A.; Bevilacqua, D.; Davidson, A.E.; Zito, I.; Thiselton, D.L.; Ressa, J.H.; Apergi, M.; et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet. 2012, 21, 3647–3654. [Google Scholar] [CrossRef] [PubMed]
- Alfano, G.; Conte, I.; Caramico, T.; Avellino, R.; Arno, B.; Pizzo, M.T.; Tanimoto, N.; Beck, S.C.; Huber, G.; Dolle, P.; et al. Vax2 regulates retinoic acid distribution and cone opsin expression in the vertebrate eye. Development 2011, 138, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Karali, M.; Peluso, I.; Marigo, V.; Banfi, S. Identification and characterization of microRNAs expressed in the mouse eye. Investig. Ophthalmol. Vis. Sci. 2007, 48, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Di Fruscio, G.; Schulz, A.; De Cegli, R.; Savarese, M.; Mutarelli, M.; Parenti, G.; Banfi, S.; Braulke, T.; Nigro, V.; Ballabio, A. Lysoplex: An efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy 2015, 11, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Savarese, M.; Di Fruscio, G.; Mutarelli, M.; Torella, A.; Magri, F.; Santorelli, F.M.; Comi, G.P.; Bruno, C.; Nigro, V. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol. Commun. 2014, 2, 100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Goode, D.L.; Cooper, G.M.; Schmutz, J.; Dickson, M.; Gonzales, E.; Tsai, M.; Karra, K.; Davydov, E.; Batzoglou, S.; Myers, R.M.; et al. Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res. 2010, 20, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mut. 2011, 32, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Tory, K.; Lacoste, T.; Burglen, L.; Moriniere, V.; Boddaert, N.; Macher, M.A.; Llanas, B.; Nivet, H.; Bensman, A.; Niaudet, P.; et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: Potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J. Am. Soc. Nephrol. 2007, 18, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Hoover-Fong, J.; Sobreira, N.; Jurgens, J.; Modaff, P.; Blout, C.; Moser, A.; Kim, O.H.; Cho, T.J.; Cho, S.Y.; Kim, S.J.; et al. Mutations in PCYT1A, encoding a key regulator of phosphatidylcholine metabolism, cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am. J. Hum. Genet. 2014, 94, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Testa, F.; Filippelli, M.; Brunetti-Pierri, R.; Di Fruscio, G.; Di Iorio, V.; Pizzo, M.; Torella, A.; Barillari, M.R.; Nigro, V.; Brunetti-Pierri, N.; et al. Mutations in the PCYT1A gene are responsible for isolated forms of retinal dystrophy. Eur. J. Hum. Genet. 2017, 25, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Sundin, O.H.; Yang, J.M.; Li, Y.; Zhu, D.; Hurd, J.N.; Mitchell, T.N.; Silva, E.D.; Maumenee, I.H. Genetic basis of total colourblindness among the Pingelapese islanders. Nat. Genet. 2000, 25, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Perrault, I.; Hanein, S.; Gerard, X.; Delphin, N.; Fares-Taie, L.; Gerber, S.; Pelletier, V.; Merce, E.; Dollfus, H.; Puech, B.; et al. Spectrum of SPATA7 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum. Mutat. 2010, 31, E1241–E1250. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Lehesjoki, A.E.; Mole, S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, A.I.; ten Brink, J.B.; de Kok, Y.J.; van Soest, S.; van den Born, L.I.; van Driel, M.A.; van de Pol, D.J.; Payne, A.M.; Bhattacharya, S.S.; Kellner, U.; et al. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat. Genet. 1999, 23, 217–221. [Google Scholar] [PubMed]
- Den Hollander, A.I.; Davis, J.; van der Velde-Visser, S.D.; Zonneveld, M.N.; Pierrottet, C.O.; Koenekoop, R.K.; Kellner, U.; van den Born, L.I.; Heckenlively, J.R.; Hoyng, C.B.; et al. CRB1 mutation spectrum in inherited retinal dystrophies. Hum. Mutat. 2004, 24, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Mears, A.J.; Gieser, L.; Yan, D.; Chen, C.; Fahrner, S.; Hiriyanna, S.; Fujita, R.; Jacobson, S.G.; Sieving, P.A.; Swaroop, A. Protein-truncation mutations in the RP2 gene in a North American cohort of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 1999, 64, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Otto, E.A.; Helou, J.; Allen, S.J.; O’Toole, J.F.; Wise, E.L.; Ashraf, S.; Attanasio, M.; Zhou, W.; Wolf, M.T.; Hildebrandt, F. Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum. Mutat. 2008, 29, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Bowne, S.J.; Sullivan, L.S.; Blackshaw, S.; Cepko, C.L.; Payne, A.M.; Bhattacharya, S.S.; Khaliq, S.; Qasim Mehdi, S.; Birch, D.G.; et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat. Genet. 2000, 24, 79–83. [Google Scholar] [PubMed]
- Valente, E.M.; Silhavy, J.L.; Brancati, F.; Barrano, G.; Krishnaswami, S.R.; Castori, M.; Lancaster, M.A.; Boltshauser, E.; Boccone, L.; Al-Gazali, L.; et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006, 38, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Marx, T.; Giddings, I.; Jagle, H.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Zrenner, E.; Sharpe, L.T.; Wissinger, B. Total colourblindness is caused by mutations in the gene encoding the α-subunit of the cone photoreceptor cGMP-gated cation channel. Nat. Genet. 1998, 19, 257–259. [Google Scholar] [PubMed]
- Lotery, A.J.; Namperumalsamy, P.; Jacobson, S.G.; Weleber, R.G.; Fishman, G.A.; Musarella, M.A.; Hoyt, C.S.; Heon, E.; Levin, A.; Jan, J.; et al. Mutation analysis of 3 genes in patients with Leber congenital amaurosis. Arch. Ophthalmol. 2000, 118, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Siemiatkowska, A.M.; van den Born, L.I.; van Genderen, M.M.; Bertelsen, M.; Zobor, D.; Rohrschneider, K.; van Huet, R.A.; Nurohmah, S.; Klevering, B.J.; Kohl, S.; et al. Novel compound heterozygous NMNAT1 variants associated with Leber congenital amaurosis. Mol. Vis. 2014, 20, 753–759. [Google Scholar] [PubMed]
- Perrault, I.; Hanein, S.; Zanlonghi, X.; Serre, V.; Nicouleau, M.; Defoort-Delhemmes, S.; Delphin, N.; Fares-Taie, L.; Gerber, S.; Xerri, O.; et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet. 2012, 44, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Halbritter, J.; Diaz, K.; Chaki, M.; Porath, J.D.; Tarrier, B.; Fu, C.; Innis, J.L.; Allen, S.J.; Lyons, R.H.; Stefanidis, C.J.; et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012, 49, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M. Leber congenital amaurosis—A model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 2007, 144, 791–811. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, M.; Khan, M.I.; Micheal, S.; Ahmed, W.; Shah, A.; Venselaar, H.; Bokhari, H.; Azam, A.; Waheed, N.K.; Collin, R.W.; et al. Identification of recurrent and novel mutations in TULP1 in Pakistani families with early-onset retinitis pigmentosa. Mol. Vis. 2012, 18, 1226–1237. [Google Scholar] [PubMed]
- Simonelli, F.; Ziviello, C.; Testa, F.; Rossi, S.; Fazzi, E.; Bianchi, P.E.; Fossarello, M.; Signorini, S.; Bertone, C.; Galantuomo, S.; et al. Clinical and molecular genetics of Leber’s congenital amaurosis: A multicenter study of Italian patients. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4284–4290. [Google Scholar] [CrossRef] [PubMed]
- Wissinger, B.; Gamer, D.; Jagle, H.; Giorda, R.; Marx, T.; Mayer, S.; Tippmann, S.; Broghammer, M.; Jurklies, B.; Rosenberg, T.; et al. CNGA3 mutations in hereditary cone photoreceptor disorders. Am. J. Hum. Genet. 2001, 69, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, X.; Zou, X.; Xu, S.; Li, H.; Soens, Z.T.; Wang, K.; Li, Y.; Dong, F.; Chen, R.; et al. Comprehensive Molecular Diagnosis of a Large Chinese Leber Congenital Amaurosis Cohort. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3642–3655. [Google Scholar] [CrossRef] [PubMed]
- Burnight, E.R.; Wiley, L.A.; Drack, A.V.; Braun, T.A.; Anfinson, K.R.; Kaalberg, E.E.; Halder, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 2014, 21, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Guan, L.; Li, S.; Zhang, J.; Xiao, X.; Jiang, H.; Yang, J.; Guo, X.; Wang, J.; Zhang, Q. Mutation analysis of Leber congenital amaurosis associated genes in patients with retinitis pigmentosa. Mol. Med. Rep. 2015, 11, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Cuzcano, A.; Koenekoop, R.K.; Coppieters, F.; Kohl, S.; Lopez, I.; Collin, R.W.; De Baere, E.B.; Roeleveld, D.; Marek, J.; Bernd, A.; et al. IQCB1 mutations in patients with leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Alasil, T.; Keane, P.A.; Sim, D.A.; Tufail, A.; Rauser, M.E. Optical coherence tomography in pediatric ophthalmology: Current roles and future directions. Ophthalmic Surg. Lasers Imaging Retin. 2013, 44, S19–S29. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Tao, Y.; Deng, W.T.; Zhu, P.; Li, J.; Dai, X.; Zhang, Y.; Shi, W.; Liu, X.; Chiodo, V.A.; et al. Vitreal delivery of AAV vectored Cnga3 restores cone function in CNGA3-/-/Nrl-/- mice, an all-cone model of CNGA3 achromatopsia. Hum. Mol. Genet. 2015, 24, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.A.; Pennesi, M.E. Retinal Gene Therapy: Current Progress and Future Prospects. Expert Rev. Ophthalmol. 2015, 10, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Muhlfriedel, R.; Tanimoto, N.; Krishnamoorthy, V.; Koch, S.; Fischer, M.D.; Becirovic, E.; Bai, L.; Huber, G.; Beck, S.C.; et al. Restoration of cone vision in the CNGA3-/- mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 2010, 18, 2057–2063. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient ‡ | Diagnosis | Gene | RefSeq | Allele 1 (nt †) | Allele 1 (prot. §) | Reference | Allele 2 (nt †) | Allele 2 (prot. §) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 6 | LCA | PCYT1A | NM_005017 | chr3:195966468; c.847C>T | p.(R283*) | [34] | chr3:195975135; c.277G>A | p.(A93T) | [35] |
| 7 | LCA | PCYT1A | NM_005017 | chr3:195966468; c.847C>T | p.(R283*) | [34] | chr3:195975135; c.277G>A | p.(A93T) | [35] |
| 8 | ACHM | CNGB3 | NM_019098 | chr8:87656009; c.1148delC | p.(T383fs) | [36] | chr8:87660049; c.970A>G | p.(R324G) | this study |
| 9 | ACHM | CNGB3 | NM_019098 | chr8:87656009; c.1148delC | p.(T383fs) | [36] | chr8:87660049; c.970A>G | p.(R324G) | this study |
| 10 | LCA | CNGB3 | NM_019098 | chr8:87656009; c.1148delC | p.(T383fs) | [36] | chr8:87645015; c.1285delT | p.(S429fs) | this study |
| 12 | LCA | SPATA7 | NM_001040428 | chr14:88893049; c.749+1G>A | p.? | [37] | chr14:88903937; c.1115A>G | p.(E372G) | this study |
| 15 | EORP | CLN3 | NM_001042432 | chr16:28497785; c.258_259del | p.(G187fs) | [38] | chr16:28497972; c.161-1G>C | p.? | [38] |
| 16 | LCA | CRB | NM_001193640 | chr1:197396689; c.1898C>T | p.(T633M) | [39] | chr1:197404419; c.3091delT | p.(C1031fs) | [40] |
| 19 | EORP | RP2 | NM_006915 | chrX:46713166; c.358C>T | p.(R120*) | [41] | - | ||
| 21 | EORP | IQCB1 | NM_001023571 | chr3:121491506; c.1066C>T | p.(R356*) | [42] | chr3:121491506; c.1066C>T | p.(R356*) | [42] |
| 22 | EORP | AIPL1 | NM_001033054 | chr17:6329101; c.645G>A | p.(W215*) | [43] | chr17:6329101; c.645G>A | p.(W215*) | [43] |
| 23 | LCA | CEP290 | NM_025114 | chr12:88490755; c.3012delA | p.(K1004fs) | this study | chr12:88477704; c.4732G>T | p.(E1578*) | [44] |
| 24 | ACHM | CNGA3 | NM_001079878 | chr2:99013274; c.1587C>A | p.(F529L) | [45] | chr2:99013274; c.1587C>A | p.(F529L) | [45] |
| 25 | LCA | GUCY2D | NM_000180 | chr17:7917236; c.2302C>T | p.(R768W) | [46] | chr17:7917236; c.2302C>T | p.(R768W) | [46] |
| 27 | LCA | NMNAT1 | NM_022787 | chr1:10032184; c.53A>G | p.(N18S) | [47] | chr1:10042461; c.542A>G | p.(Y181C) | [48] |
| 29 | LCA | PCYT1A | NM_005017 | chr3:195966417; c.897+1G>A | p.? | [34] | chr3:195975135; c.277G>A | p.(A93T) | [35] |
| 31 | EORP | CEP290 | NM_025114 | chr12:88449397; c.6916A>T | p.(R2306*) | this study | chr12:88508262; c.1987A>T | p.(K663*) | [49] |
| 32 | EORP | IQCB1 | NM_001023570 | chr3:121527767; c.479_482del | p.(I160fs) | this study | chr3:121515964; c.876+1G>T | p.? | this study |
| 33 | LCA | CRB1 | NM_001193640 | chr1:197237597; c.55_56insT | p.(L19fs) | [50] | chr1:197391051; c.1757G>A | p.(C586Y) | this study |
| 36 | EORP | TULP1 | NM_001289395 | chr6:35467808; c.1286G>A | p.(R429Q) | [51] | chr6:35467808; c.1286G>A | p.(R429Q) | [51] |
| 37 | EORP | CRB1 | NM_001193640 | chr1:197390271; c.977G>A | p.(C326Y) | [52] | chr1:197390271; c.977G>A | p.(C326Y) | [52] |
| 39 | ACHM | PDE6C | NM_006204 | chr10:95415598; c.2017G>T | p.(D673Y) | this study | chr10:95415598; c.2017G>T | p.(D673Y) | this study |
| 40 | ACHM | CNGA3 | NM_001079878 | chr2:99012747; c.1060C>T | p.(P354S) | [53] | chr2:99012747; c.1060C>T | p.(P354S) | [53] |
| 41 | LCA | RPGRIP1 | NM_020366 | chr14:21762833; c.86-3T>G | p.? | this study | chr14:21793399; c.2225_2226del | p.(G742fs) | this study |
| 42 | LCA | GUCY2D | NM_000180 | chr17:7912823; c.1669-1G>A | p.? | this study | chr17:7912823; c.1669-1G>A | p.? | this study |
| Patient | Age | Age of Onset | Nystagmus | BCVA † RE/LE ‡ | Fundus | MT § (μm) RE/LE ‡ | EZ Band ¶ | ERG # Scotopic RE/LE ‡ (μV) | ERG # Photopic RE/LE ‡ (μV) | Mutated Gene |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 y | 3 m | yes | n.a. | “salt & pepper” | n.a. | n.a. | u.n.l. | u.n.l. | - |
| 2 | 5 y | 6 m | yes | n.a. | “salt & pepper” | 229/220 | absent | n.a. | n.a. | - |
| 3 | 4 y | 4 m | yes | n.a. | “salt & pepper” | n.a. | n.a. | u.n.l. | u.n.l. | - |
| 4 | 17 y | 6 m | yes | HM/HM | “salt & pepper” | 252/119 | irregular | 15/34.9 | 53.6/25.0 | - |
| 5 | 14 y | 3 m | no | HM/HM | RPE dystrophy | 179/181 | irregular | u.n.l. | u.n.l. | - |
| 6 | 18 y | 8 m | yes | 0.2/0.02 | RP | 176/120 | absent | u.n.l. | u.n.l. | PCYT1A |
| 7 | 10 y | 1 m | yes | 0.05/0.05 | RP | 103/103 | absent | 18.9/25.2 | 32.6/20.6 | PCYT1A |
| 10 | 5 y | 4 m | yes | 0.1/0.1 | normal | 168/156 | irregular | u.n.l. | u.n.l. | CNGB3 |
| 11 | 16 y | 9 m | yes | 0.05/0.05 | RPE dystrophy | n.a. | n.a. | 26.2/39.1 | 27.8/24.28 | - |
| 12 | 6 y | 7 m | yes | LP/LP | “salt & pepper” | 260/219 | absent | u.n.l. | u.n.l. | SPATA7 |
| 14 | 9 y | 6 m | yes | n.a. | “salt & pepper” | n.a. | n.a. | n.a. | n.a. | CEP290 ? |
| 16 | 5 y | 3 m | yes | 0.1/0.1 | RPE dystrophy | 237/328 | absent | u.n.l. | u.n.l. | CRB1 |
| 17 | 17 y | 2 m | yes | HM/HM | “salt & pepper” | 149/104 | absent | u.n.l. | u.n.l. | - |
| 23 | 3 y | 1 m | yes | n.a. | “salt & pepper” | n.a. | n.a. | n.a. | n.a. | CEP290 |
| 25 | 2 y | 4 m | yes | n.a. | normal | n.a. | n.a. | n.a. | n.a. | GUCY2D |
| 26 | 8 y | 2 m | yes | LP/LP | “salt & pepper” | n.a. | n.a. | n.a. | n.a. | CEP290 ? |
| 27 | 5 y | 9 m | yes | n.a. | “salt & pepper” | n.a. | n.a. | n.a. | n.a. | NMNAT1 |
| 29 | 3 y | 4 m | yes | 0.1/0.1 | “salt & pepper” | 110/100 | absent | u.n.l. | u.n.l. | PCYT1A |
| 30 | 11 y | 3 m | yes | LP/LP | RPE dystrophy | n.a. | n.a. | u.n.l. | u.n.l. | - |
| 33 | 5 y | 3 m | yes | 0.1/0.1 | “salt & pepper” | n.a. | n.a. | n.a. | n.a. | CRB1 |
| 34 | 7 y | 1 m | yes | LP/LP | “salt & pepper” | 170/146 | absent | u.n.l. | u.n.l. | CEP290 ? |
| 38 | 13 y | 9 m | yes | 0.03/0.02 | “salt & pepper” | n.a. | n.a. | u.n.l. | u.n.l. | - |
| 41 | 8 y | 1 m | yes | 0.05/0.05 | “salt & pepper” | 243/242 | irregular | u.n.l. | u.n.l. | RPGRIP1 |
| 42 | 2 y | 1 m | yes | n.a. | normal | n.a. | n.a. | n.a. | n.a. | GUCY2D |
| Patient | Age | Age of Onset | Nystagmus | BCVA † RE/LE ‡ | Fundus | MT § (µm) RE/LE ‡ | EZ Band ¶ | ERG # Scotopic RE/LE ‡ (μV) | ERG # Photopic RE/LE ‡ (μV) | Mutated Gene |
|---|---|---|---|---|---|---|---|---|---|---|
| 13 | 16 y | 2 y | no | 0.2/0.2 | RPE dystrophy | 101/105 | absent | u.n.l. | u.n.l. | - |
| 15 | 11 y | 2 y | no | HM/HM | RPE dystrophy | 154/142 | absent | n.a. | n.a. | CLN3 |
| 18 | 16 y | 2 y | no | 0.3/0.3 | “salt & pepper” | 205/197 | irregular | 61.4/69.2 | 55.5/62.1 | - |
| 19 | 18 y | 8 m | yes | 0.1/0.1 | RPE dystrophy | 114/157 | absent | u.n.l. | 26.1/13.4 | RP2 |
| 20 | 7 y | 2 y | no | 0.3/0.3 | RPE dystrophy | 126/119 | absent | u.n.l. | u.n.l. | - |
| 21 | 8 y | 9 m | yes | 0.3/0.3 | RPE dystrophy | 243/247 | absent | u.n.l. | u.n.l. | IQCB1 |
| 22 | 7 y | 9 m | yes | 0.008/0.008 | “salt & pepper” | 129/114 | absent | 11.6/1.14 | 4.96/4.66 | AIPL1 |
| 28 | 2 y | 8 m | yes | n.a. | normal | n.a. | n.a. | n.a. | n.a. | - |
| 31 | 8 y | 9 m | yes | 0.2/0.2 | “salt & pepper” | 168/173 | irregular | u.n.l. | u.n.l. | CEP290 |
| 32 | 10 y | 2 y | no | 0.05/0.05 | “salt & pepper” | 180/213 | absent | 14.8/25 | 12.2/1.22 | IQCB1 |
| 35 | 11 y | 9 m | yes | 0.2/0.2 | normal | 137/170 | irregular | u.n.l. | u.n.l. | - |
| 36 | 16 y | 2 y | no | 0.05/0.05 | “salt & pepper” | 192/215 | absent | u.n.l. | u.n.l. | TULP1 |
| 37 | 5 y | 1 y | no | 0.2/0.3 | RPE dystrophy | 101/104 | absent | u.n.l. | u.n.l. | CRB1 |
| 43 | 11 y | 8 m | no | 0.2/0.2 | RP | 165/180 | absent | u.n.l. | u.n.l. | - |
| Patient | Age | Age of Onset | Nystagmus | BCVA † RE/LE ‡ | Fundus | MT § (μm) RE/LE ‡ | EZ Band ¶ | ERG # Scotopic RE/LE ‡ (μV) | ERG # Photopic RE/LE ‡ (μV) | Mutated Gene |
|---|---|---|---|---|---|---|---|---|---|---|
| 8 | 12 y | 1 y | yes | 0.1/0.1 | pigment mottling | 258/294 | disruption | n.a. | n.a. | CNGB3 |
| 9 | 13 y | 2 y | yes | 0.1/0.1 | pigment mottling | 166/168 | disruption | 128/148 | 7.45/13.1 | CNGB3 |
| 24 | 10 y | 1 y | yes | 0.2/0.1 | Normal | 188/175 | irregular | u.n.l. | u.n.l. | CNGA3 |
| 39 | 16 y | 1 y | yes | 0.2/0.2 | pigment mottling | 148/142 | disruption | u.n.l. | u.n.l. | PDE6C |
| 40 | 12 y | 1 y | yes | 0.2/0.2 | Normal | 149/128 | irregular | 61.6/81.9 | 50.9/63.2 | CNGA3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Iorio, V.; Karali, M.; Brunetti-Pierri, R.; Filippelli, M.; Di Fruscio, G.; Pizzo, M.; Mutarelli, M.; Nigro, V.; Testa, F.; Banfi, S.; et al. Clinical and Genetic Evaluation of a Cohort of Pediatric Patients with Severe Inherited Retinal Dystrophies. Genes 2017, 8, 280. https://doi.org/10.3390/genes8100280
Di Iorio V, Karali M, Brunetti-Pierri R, Filippelli M, Di Fruscio G, Pizzo M, Mutarelli M, Nigro V, Testa F, Banfi S, et al. Clinical and Genetic Evaluation of a Cohort of Pediatric Patients with Severe Inherited Retinal Dystrophies. Genes. 2017; 8(10):280. https://doi.org/10.3390/genes8100280
Chicago/Turabian StyleDi Iorio, Valentina, Marianthi Karali, Raffaella Brunetti-Pierri, Mariaelena Filippelli, Giuseppina Di Fruscio, Mariateresa Pizzo, Margherita Mutarelli, Vincenzo Nigro, Francesco Testa, Sandro Banfi, and et al. 2017. "Clinical and Genetic Evaluation of a Cohort of Pediatric Patients with Severe Inherited Retinal Dystrophies" Genes 8, no. 10: 280. https://doi.org/10.3390/genes8100280
APA StyleDi Iorio, V., Karali, M., Brunetti-Pierri, R., Filippelli, M., Di Fruscio, G., Pizzo, M., Mutarelli, M., Nigro, V., Testa, F., Banfi, S., & Simonelli, F. (2017). Clinical and Genetic Evaluation of a Cohort of Pediatric Patients with Severe Inherited Retinal Dystrophies. Genes, 8(10), 280. https://doi.org/10.3390/genes8100280