
Development of Novel Polymorphic EST-SSR Markers in Bailinggu (Pleurotus tuoliensis) for Crossbreeding


Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Protoplast-Derived Monokaryons and Determination of Mating Types
2.2. Isolation of Basidiospore-Derived Monokaryons
2.3. Crossbreeding of Monokaryons
2.4. Development of Novel Expressed Sequence Tag-Simple Sequence Repeat Markers Using Transcriptome Sequencing
2.5. Validation of Novel Expressed Sequence Tag-Simple Sequence Repeat Markers
2.6. Across-Taxa Transferability of Novel Expressed Sequence Tag-Simple Sequence Repeat Markers in Monokaryons of Related Species of the Genus Pleurotus
3. Results and Discussion
3.1. Isolation of Protoplast-Derived Monokaryons
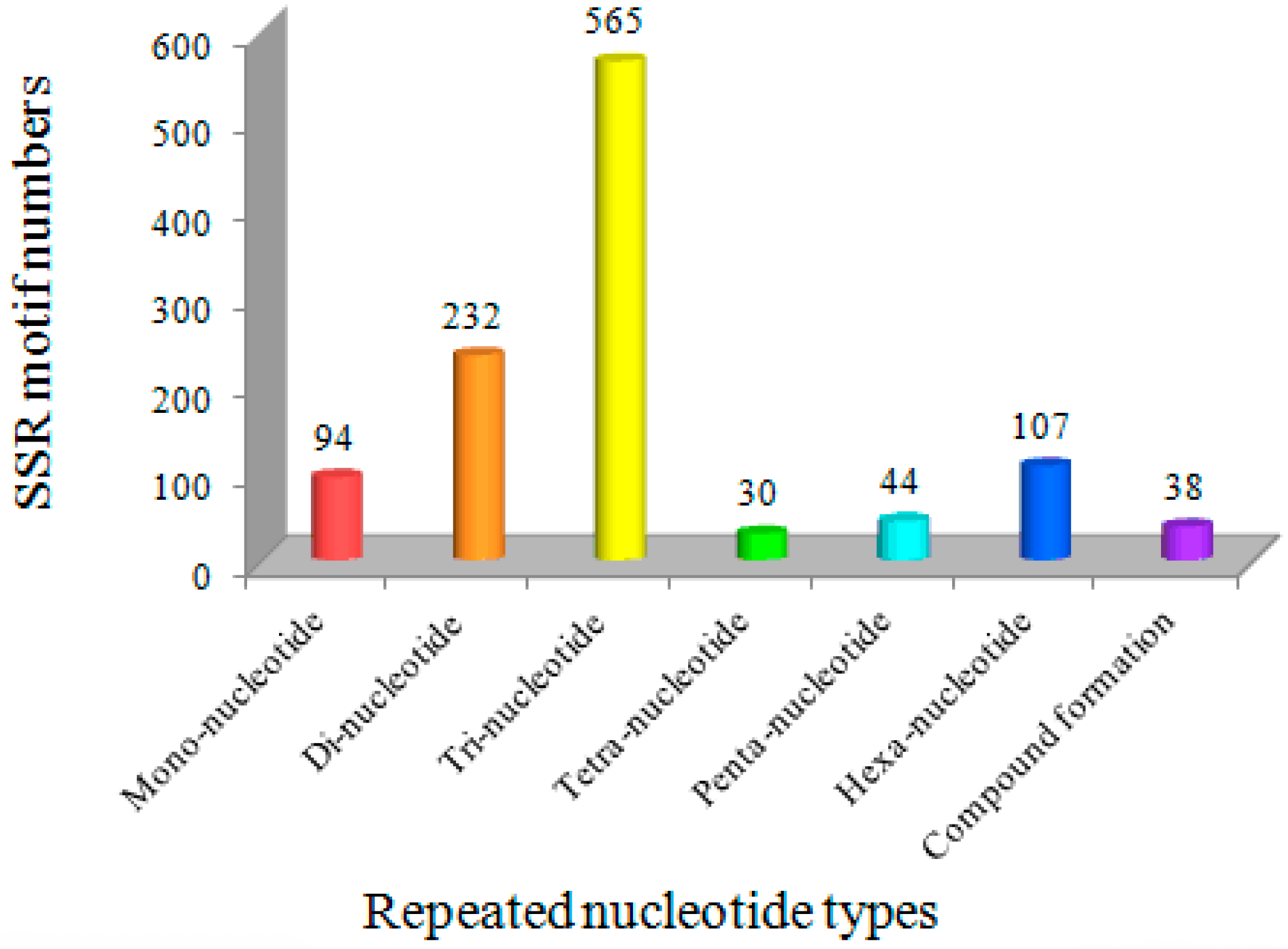
3.2. Novel EST-SSR Markers Developed Using Transcriptome Sequencing
3.3. Validation of Novel Expressed Sequence Tag-Simple Sequence Repeats
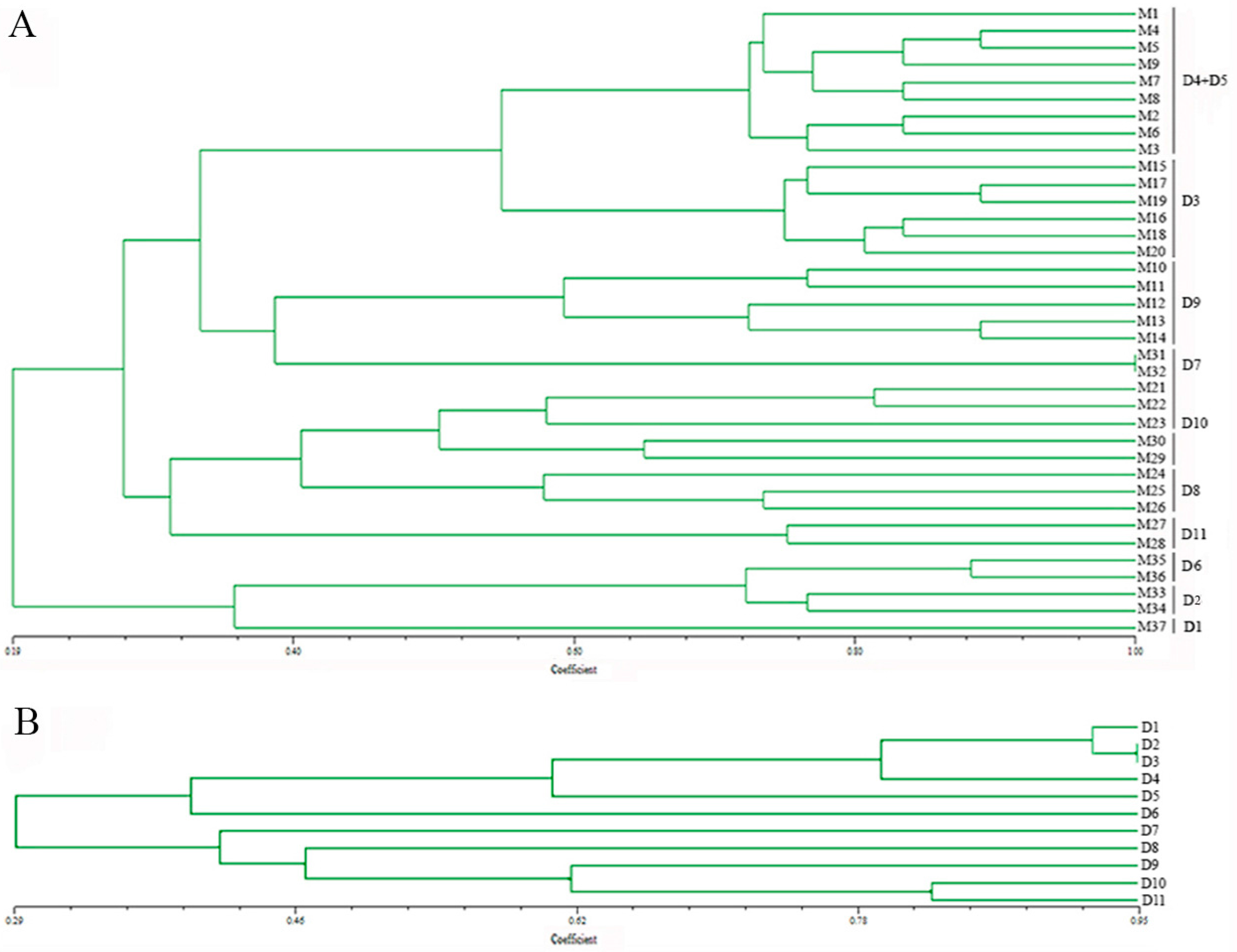
3.4. Genetic Diversity Analysis of Monokaryons and Dikaryons Using 18 EST-SSRs
3.5. Across-Taxa Transferability of Novel Expressed Sequence Tag-Simple Sequence Repeat Markers in the Monokaryons of Two Species of the Genus Pleurotus
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhao, M.R.; Zhang, J.X.; Chen, Q.; Wu, X.L.; Gao, W.; Deng, W.Q.; Huang, C.Y. The famous cultivated mushroom Bailinggu is a separate species of the Pleurotus eryngii species complex. Sci. Rep. 2016, 6, 33066. [Google Scholar] [CrossRef] [PubMed]
- Kawai, G.; Babasaki, K.; Neda, H. Taxonomic position of a Chinese Pleurotus “Bai-Ling-Gu”, it belongs to Pleurotus eryngii (DC., Fr.) Quél. and evolved independently in China. Mycoscience 2008, 49, 75–87. [Google Scholar] [CrossRef]
- Mou, C.J.; Cao, Y.Q.; Ma, J.L. A new variety of Pleurotus eryngii and its cultural characters. Acta Mycol. Sin. 1987, 6, 153–156. [Google Scholar]
- Zervakis, G.I.; Ntougias, S.; Gargano, M.L.; Besi, M.I.; Polemis, E.; Typas, M.A.; Venturella, G. A reappraisal of the Pleurotus eryngii complex—New species and taxonomic combinations based on the application of a polyphasic approach, and an identification key to Pleurotus taxa associated with Apiaceae plants. Fungal Biol. 2014, 118, 814–834. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.P.; Yueting Dai, Y.T.; Yang, C.T.; Wei, P.; Song, B.; Yang, Y.; Sun, L.; Zhang, Z.W.; Li, Y. Comparative transcriptome analysis identified candidate genes related to Bailinggu mushroom formation and genetic markers for genetic analyses and breeding. Sci. Rep. 2017, 7, 9266. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Ryu, J.-S.; Lee, Y.-H.; Kim, H.-R. Breeding of a long shelf-life strain for commercial cultivation by mono–mono crossing in Pleurotus eryngii. Sci. Hortic. 2013, 162, 265–270. [Google Scholar] [CrossRef]
- Liu, S.R.; Ke, B.B.; Zhang, W.R.; Liu, X.H.; Wu, X.P. Breeding of new Ganoderma lucidum strains simultaneously rich in polysaccharides and triterpenes by mating basidiospore-derived monokaryons of two commercial cultivars. Sci. Hortic. 2017, 216, 58–65. [Google Scholar] [CrossRef]
- Kim, K.-H.; Kang, Y.M.; Im, C.H.; Ali, A.; Kim, S.Y.; Je, H.-J.; Kim, M.-K.; Rho, H.S.; Lee, H.S.; Kong, W.-S.; et al. Identification and functional analysis of pheromone and receptor genes in the B3 mating locus of Pleurotus eryngii. PLoS ONE 2014, 9, e104693. [Google Scholar]
- Li, D.; Liu, Y.; Wang, P.; Ma, Y.; Wang, S.; Zhao, S.; Xu, F. Development of SCAR markers to determine the mating types of Lepista nuda protoplast monokaryons. Curr. Microbiol. 2014, 68, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Feng, B.; Li, J.; Yan, C.; Yang, Z.L. Genetic diversity and breeding history of winter mushroom (Flammulina velutipes) in China uncovered by genomic SSR markers. Gene 2016, 591, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jung, J.; Kim, M.-S.; Lee, J.M.; Choi, D.; Yeam, I. Molecular marker development and genetic diversity exploration by RNA-seq in Platycodon grandiflorum. Genome 2015, 58, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.Y.; Hu, D.D.; Gu, J.G.; Hu, Q.X.; Zuo, X.M.; Wang, H.X. Development of SSR markers for typing cultivars in the mushroom Auricularia auricula-judae. Mycol. Prog. 2012, 11, 587–592. [Google Scholar] [CrossRef]
- Yan, H.D.; Zhang, Y.; Zeng, B.; Yin, G.H.; Zhang, X.Q.; Ji, Y.; Huang, L.K.; Jiang, X.M.; Liu, X.C.; Peng, Y.; et al. Genetic diversity and association of EST-SSR and SCoT markers with rust traits in orchardgrass (Dactylis glomerata L.). Molecules 2016, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, X.; Jian, D.; Zhan, Y.; Fan, G. De novo transcriptome analysis of a medicinal fungi Phellinus linteus and identification of SSR markers. Biotechnol. Biotechnol. Equip. 2015, 29, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Raveendar, S.; Suresh, S.; Lee, G.A.; Lee, J.R.; Cho, J.H.; Lee, S.Y.; Ma, K.H.; Cho, G.T.; Chung, J.W. Transcriptome analysis of two Vicia sativa subspecies: Mining molecular markers to enhance genomic resources for vetch improvement. Genes 2015, 6, 1164–1182. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.P.; Liang, Y.; Dai, Y.T.; Yang, C.T.; Duan, M.Z.; Zhang, Z.; Hu, S.N.; Zhang, Z.W.; Li, Y. De novo sequencing and transcriptome analysis of Pleurotus eryngii subsp. tuoliensis (Bailinggu) mycelia in response to cold stimulation. Molecules 2016, 21, 560. [Google Scholar]
- Wang, M.Z.; Liu, S.S.; Li, Y.Y.; Xu, R.; Lu, C.H.; Shen, Y.M. Protoplast mutation and genome shuffling induce the endophytic fungus Tubercularia sp. TF5 to produce new compounds. Curr. Microbiol. 2010, 61, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.N.; Casaletti, L.; Bao, S.N.; Borges, C.L.; Lima, P.D.S.; Soares, C.M.D.A. Characterizing the nuclear proteome of Paracoccidioides spp. Fungal Biol. 2016, 120, 1209e1224. [Google Scholar] [CrossRef] [PubMed]
- Larraya, L.; Peñas, M.M.; Pérez, G.; Santos, C.; Ritter, E.; Pisabarro, A.G.; Ramírez, L. Identification of incompatibility alleles and characterisation of molecular markers genetically linked to the A incompatibility locus in the white rot fungus Pleurotus ostreatus. Curr. Genet. 1999, 34, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.M.; Li, J.K.; Xiao, S.J.; Liu, X.D. De novo assembly and characterization of foot transcriptome and microsatellite marker development for Paphia textile. Gene 2016, 576, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Parekha, M.J.; Kumar, S.; Zalaa, H.N.; Fougat, R.S.; Patel, C.B.; Bosamia, T.C.; Kulkarni, K.S.; Parihar, A. Development and validation of novel fiber relevant dbEST-SSR markers and their utility in revealing genetic diversity in diploid cotton (Gossypium herbaceum and G. arboreum). Ind. Crop. Prod. 2016, 83, 620–629. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, S.T. Monokaryotization by protoplasting heterothallic species of edible mushrooms. World J. Microbiol. Biotechnol. 1993, 9, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.C.; Zhang, S.T. Analysis of incompatibility factors in cultivated strains of Lentinus edodes in China. J. Huazhong Agric. Univ. 1995, 14, 459–466. [Google Scholar]
- Seong, J.; Kang, S.W.; Patnaik, B.B.; Park, S.Y.; Hwang, H.J.; Chung, J.M.; Song, D.K.; Noh, M.Y.; Park, S.-H.; Jeon, G.J.; et al. Transcriptome analysis of the tadpole shrimp (Triops longicaudatus) by Illumina paired-end sequencing: Assembly, annotation, and marker discovery. Genes 2016, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Vatanparast, M.; Shetty, P.; Chopra, R.; Doyle, J.J.; Sathyanarayana, N.; Egan, A.N. Transcriptome sequencing and marker development in winged bean (Psophocarpus tetragonolobus; Leguminosae). Sci. Rep. 2016, 6, 29070. [Google Scholar] [CrossRef] [PubMed]
- Foulongne-Oriol, M.; Lapalu, N.; Ferandon, C.; Spataro, C.; Ferrer, N.; Amselem, J.; Savoie, J.M. The first set of expressed sequence tags (EST) from the medicinal mushroom Agaricus subrufescens delivers resource for gene discovery and marker development. Appl. Microbiol. Biol. 2014, 98, 7879–7892. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.D.; Lu, N.; Chen, Q.; Song, J.L.; Wang, W.K. Analysis on transcriptome sequenced of maitake. J. Fudan Univ. 2015, 54, 673–678. [Google Scholar]
- Zhou, Y.; Chen, L.F.; Fan, X.Z.; Bian, Y.B. De novo assembly of Auricularia polytricha transcriptome using Illumina sequencing for gene discovery and SSR marker identification. PLoS ONE 2014, 9, e91740. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.X.; Cheng, S.M.; Li, A.Z.; Lin, F.C. Compose and distribution of EST-SSR in genomes of Lentinula edodes. J. Microbiol. Chin. 2007, 34, 438–442. [Google Scholar]
- Wang, Y.; Chen, M.J.; Wang, H.; Bao, D.P. Distribution pattern analysis of SSR loci in the genome of Boletus edulis. Mycosystema 2015, 34, 204–214. [Google Scholar]
- Wang, Y.; Chen, M.; Wang, H.; Wang, J.F.; Bao, D. Microsatellites in the genome of the edible mushroom, Volvariella volvacea. Biol. Med. Res. Int. 2014, 2014, 281912. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yan, H.D.; Zhang, X.Q.; Zhang, J.; Frazier, T.P.; Huang, D.J.; Lu, L.; Huang, L.K.; Liu, W.; Peng, Y.; et al. De novo transcriptome analysis and molecular marker development of two Hemarthria species. Front. Plant Sci. 2016, 7, 496. [Google Scholar] [CrossRef] [PubMed]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72. [Google Scholar] [PubMed]
- Hamarsheh, O.; Amro, A. Characterization of simple sequence repeats (SSRs) from Phlebotomus papatasi (Diptera: Psychodidae) expressed sequence tags (ESTs). Parasite Vector 2011, 4, 189. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Hu, Z.; Liu, W.; Li, J.; Wang, W.; Liang, Z.; Wang, F.; Sun, X. Distribution, function and evolution characterization of microsatellite in Sargassum thunbergii (Fucales, Phaeophyta) transcriptome and their application in marker development. Sci. Rep. 2016, 6, 18947. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pandey, B.; Danishuddin, M.; Sheoran, S.; Sharma, P.; Chatrath, R. Mining and survey of simple sequence repeats in wheat rust Puccinia sp. Bioinformation 2011, 7, 291–295. [Google Scholar] [CrossRef] [PubMed]
- You, C.Y.; Chen, Q.J.; Qu, Y.U.; Ma, L. Construction and evaluation on crossing population from Gossypium hirsutum and G. barbadense. J. Xinjiang Agric. Univ. 2007, 30, 68–71. [Google Scholar]
- Zhao, Y.; Lin, F.; Yan, S.Y.; Wang, H.; Chen, M.J. Establishment and application of an efficient cross breeding method assisted by molecular markers of Volvariella volvacea. Microbiol. Chin. 2015, 42, 1165–1174. [Google Scholar]
- Xie, B.G.; Yi, H.; Huang, Z.L.; Lin, Y.Z.; Xie, F.Q.; Iiang, Y.J. Cross-breeding and isoenzyme analysis of Volvariella volvacea. J. Fujian Agric. Univ. 2001, 30, 372–376. [Google Scholar]
- Bian, Y.B.; Wu, K.Y.; Wu, C.S. Genetic analysis of isozyme loci of intraspecific hybrid in Auricularia auricula. Acta Genet. Sin. 2003, 30, 76–80. [Google Scholar] [PubMed]
- Bian, Y.B.; Luo, X.C.; Zhou, Q. Esterase isozyme zymogram polymorphisms of cultivated strains in Auricularia auricula. Mycosystema 2000, 19, 87–90. [Google Scholar]
- Wu, P.; Zhang, L.J.; Zhang, D.; Shang, X.D.; Tan, Q.; Song, C.Y. Identification of Xianggu (Lentinula edodes) monokaryons and hybrid progenies using SSR markers. Microbiol. Chin. 2016, 43, 444–455. [Google Scholar]
- Yuan, F.; Yu, H.; Zuo, S.M.; Adams, A. Development of EST-SSR for preliminary analysis of genetic diversity of Cordyceps militaris. Biochem. Syst. Ecol. 2015, 58, 126–131. [Google Scholar] [CrossRef]
- Zhang, R.; Hu, D.; Zhang, J.; Zuo, X.; Jiang, R.; Wang, H.; Ng, T.B. Development and characterization of simple sequence repeat (SSR) markers for the mushroom Flammulina velutipes. J. Biosci. Bioeng. 2010, 110, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.P.; Wang, X.X.; Li, D.; Liu, Y.; Song, B.; Zhang, C.L.; Wang, Q.; Chen, M.Y.; Zhang, Z.W.; Li, Y. Identification of resistance to wet bubble disease and genetic diversity in wild and cultivated strains of Agaricus bisporus. Int. J. Mol. Sci. 2016, 17, 1568. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Shangguan, L.F.; Song, C.N.; Wang, C.; Gao, Z.H.; Yu, H.P.; Fang, J.G. Analysis of expressed sequence tags from Prunus mume flower and fruit and development of simple sequence repeat markers. BMC Genet. 2010, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.; Budak, H.; Varshney, R.V.; Dorado, G.; Graner, A.; Hernandez, P. Transferability and polymorphism of barley EST-SSR markers used for phylogenetic analysis in Hordeum chilense. BMC Plant Biol. 2008, 8, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| No. | Strain Number | Strain Name | Strain Type | Origin |
|---|---|---|---|---|
| 1 | CCMJ814 | P. tuoliensis | Wild strain | Xinjiang, China |
| 2 | CCMJ973 | P. tuoliensis | Wild strain | Xinjiang, China |
| 3 | CCMJ980 | P. tuoliensis | Wild strain | Xinjiang, China |
| 4 | CCMJ1077 | P. tuoliensis | Cultivated strain | Xinjiang, China |
| 5 | CCMJ1123 | P. tuoliensis | Cultivated strain | Shandong, China |
| No. | Strain Number | Strain Name | Strain Type | Origin |
|---|---|---|---|---|
| 1 | CCMJ967 | P. tuoliensis | Wild strain | Xinjiang, China |
| 2 | CCMJ968 | P. tuoliensis | Wild strain | Xinjiang, China |
| 3 | CCMJ973 | P. tuoliensis | Wild strain | Xinjiang, China |
| 4 | CCMJ974 | P. tuoliensis | Wild strain | Xinjiang, China |
| 5 | CCMJ1001 | P. tuoliensis | Wild strain | Xinjiang, China |
| 6 | CCMJ1002 | P. tuoliensis | Wild strain | Xinjiang, China |
| 7 | CCMJ1044 | P. tuoliensis | Cultivated strain | Beijing, China |
| 8 | CCMJ970 | P. eryngii var. ferulae | Wild strain | Xinjiang, China |
| 9 | CCMJ1080 | P. ostreatus | Cultivated strain | Changchun, China |
| Items | Number |
|---|---|
| Total raw data | 199.81 million |
| Total clean data | 177.98 million |
| Total number of assembled unigenes | 31,139 |
| Total size of examined sequences (bp) | 60,265,141 |
| Total number of identified SSRs | 1110 |
| Number of sequences containing SSRs | 2211 |
| Number of sequences containing more than one SSR | 165 |
| Locus | Motif | Primer Sequence (5′-3′) | Na | Ne | PIC | I |
|---|---|---|---|---|---|---|
| blgSSR11 | (CCAGGA)11 | F-CTTGAATATTGACGGGAGCC R-AGGCTGTGGATAAGAACCCC | 4 | 2.603 | 0.555 | 1.011 |
| blgSSR13 | (CCA)5(CAC)6(CCT)6 | F-TCCATAATTGTCATCTCCCCA R-AAATAATTACGACGGTGGGC | 5 | 3.802 | 0.719 | 1.457 |
| blgSSR14 | (AT)7 | F-GTCAAACTGCCCAAATTCGT R-CACGGTTGCTCGATTATTCTG | 6 | 2.901 | 0.612 | 1.034 |
| blgSSR16 | (AC)7 | F-ACTGCGTCGTGGTGTACAAG R-GTCGCTAAGTATCGGTGGGA | 4 | 2.042 | 0.607 | 0.616 |
| blgSSR20 | (AG)7 | F-CGGTGCCAGTGTTCCTTATT R-TGATTCCTGCCCTTTCATTC | 5 | 2.344 | 0.413 | 0.708 |
| blgSSR24 | (GA)7 | F-ATCGGGAATGGCAATCAATA R-CTCGAGTCCCGAAACCAATA | 7 | 4.872 | 0.766 | 1.586 |
| blgSSR26 | (TCC)6 | F-CCCCGTCCTCTAGTTCATCA R-AATACGGGTCGTCAGATTCG | 5 | 3.044 | 0.682 | 1.166 |
| blgSSR27 | (GGA)8 | F-ATCGAAAAATGATTACGCCG R-GGCGAATTTCCTCTTTAGGG | 4 | 2.201 | 0.602 | 0.765 |
| blgSSR28 | (GCG)6 | F-ACTTTCCAACACCAACTGCC R-GACCACGAGAGTATCGGAGC | 4 | 3.276 | 0.650 | 1.208 |
| blgSSR29 | (GAG)7 | F-TGTTGCAAGCAATGGAATGT R-GTGAATTCCAGCGGTTGTTT | 5 | 3.293 | 0.646 | 1.247 |
| blgSSR30 | (ACC)6 | F-GAAGAGTCGCAGACCTCCAC R-GATGCTTTGGTGGACTTGGT | 3 | 1.995 | 0.431 | 0.83 |
| blgSSR31 | (CGC)5 | F-AACATGTTCTCCAAGGCCAC R-ATGCGTGCTAACTGAACGTG | 4 | 2.222 | 0.494 | 0.844 |
| blgSSR32 | (ATC)6 | F-GCGGATAACCTACTCGTGGA R-ACGCCGAAAATTTTGATGTC | 5 | 3.006 | 0.612 | 1.184 |
| blgSSR37 | (CT)6 | F-GAGCCCAAGTGACGTTCCTA R-CTAGGTGGGACTCCGAAACC | 5 | 2.377 | 0.603 | 0.993 |
| blgSSR38 | (AGC)6 | F-ATAACAAGGCATGTTTCCGC R-ACAGTCAGGCTCTGGGAGAA | 5 | 3.551 | 0.690 | 1.309 |
| blgSSR43 | (TCC)5 | F-CTCAGCCCCAAATTGAACAT R-CTAGTGGCGGGAAGACTGAG | 4 | 2.333 | 0.584 | 1.035 |
| blgSSR56 | (CCA)6 | F-GTGTGGGCAGTCGTAGCATA R-CCCTTCCGTTGCTTTCATTA | 6 | 20987 | 0.525 | 1.024 |
| blgSSR57 | (TCC)6 | F-CTCGACTCCACGAAAGAAGG R-AACACCCGAAATACGAATGC | 4 | 1.734 | 0.321 | 0.635 |
| Mean | 4.778 | 2.805 | 0.584 | 1.036 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Y.; Su, W.; Yang, C.; Song, B.; Li, Y.; Fu, Y. Development of Novel Polymorphic EST-SSR Markers in Bailinggu (Pleurotus tuoliensis) for Crossbreeding. Genes 2017, 8, 325. https://doi.org/10.3390/genes8110325
Dai Y, Su W, Yang C, Song B, Li Y, Fu Y. Development of Novel Polymorphic EST-SSR Markers in Bailinggu (Pleurotus tuoliensis) for Crossbreeding. Genes. 2017; 8(11):325. https://doi.org/10.3390/genes8110325
Chicago/Turabian StyleDai, Yueting, Wenying Su, Chentao Yang, Bing Song, Yu Li, and Yongping Fu. 2017. "Development of Novel Polymorphic EST-SSR Markers in Bailinggu (Pleurotus tuoliensis) for Crossbreeding" Genes 8, no. 11: 325. https://doi.org/10.3390/genes8110325
APA StyleDai, Y., Su, W., Yang, C., Song, B., Li, Y., & Fu, Y. (2017). Development of Novel Polymorphic EST-SSR Markers in Bailinggu (Pleurotus tuoliensis) for Crossbreeding. Genes, 8(11), 325. https://doi.org/10.3390/genes8110325