Dynamics of p53: A Master Decider of Cell Fate


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
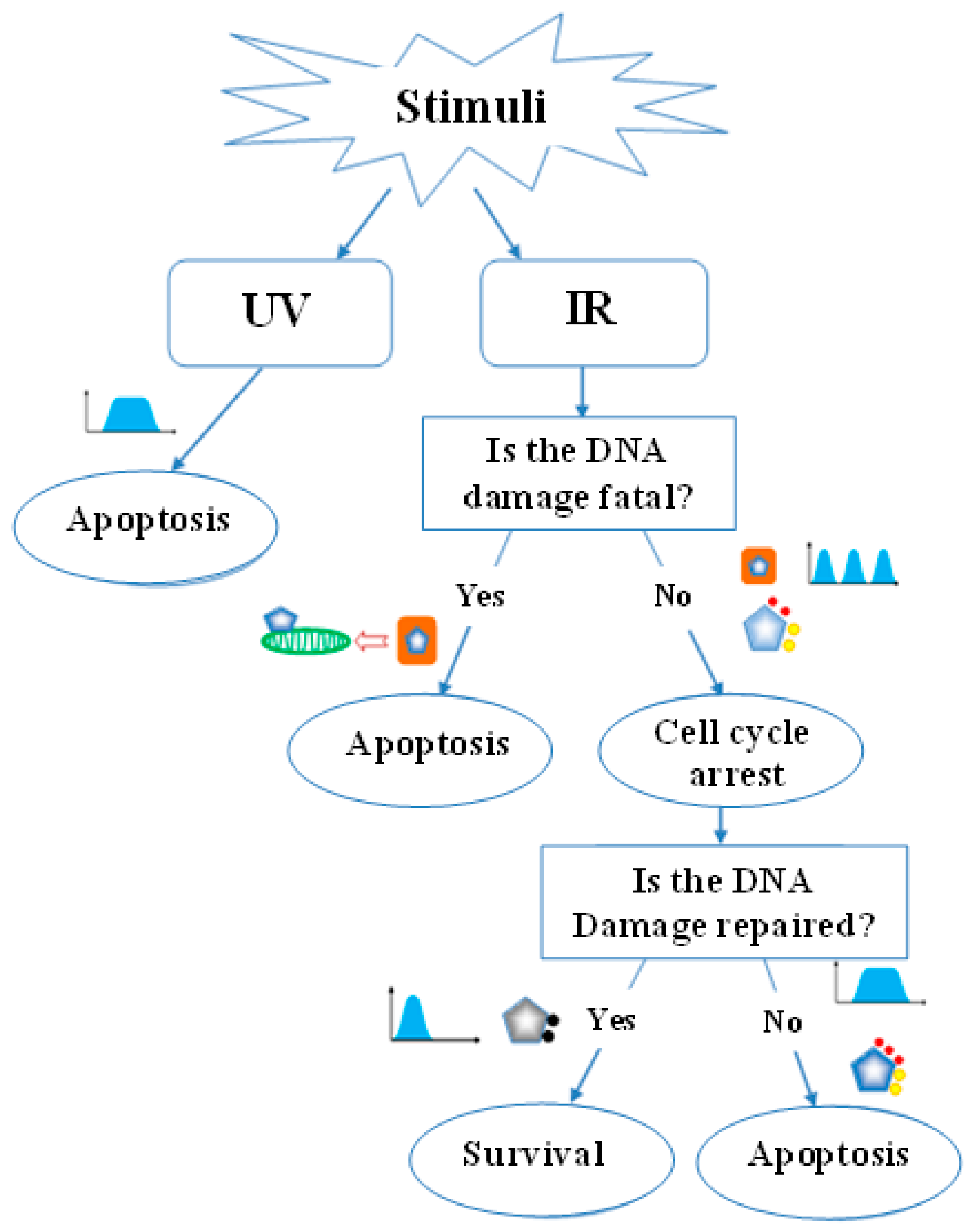
Abstract
:1. Introduction
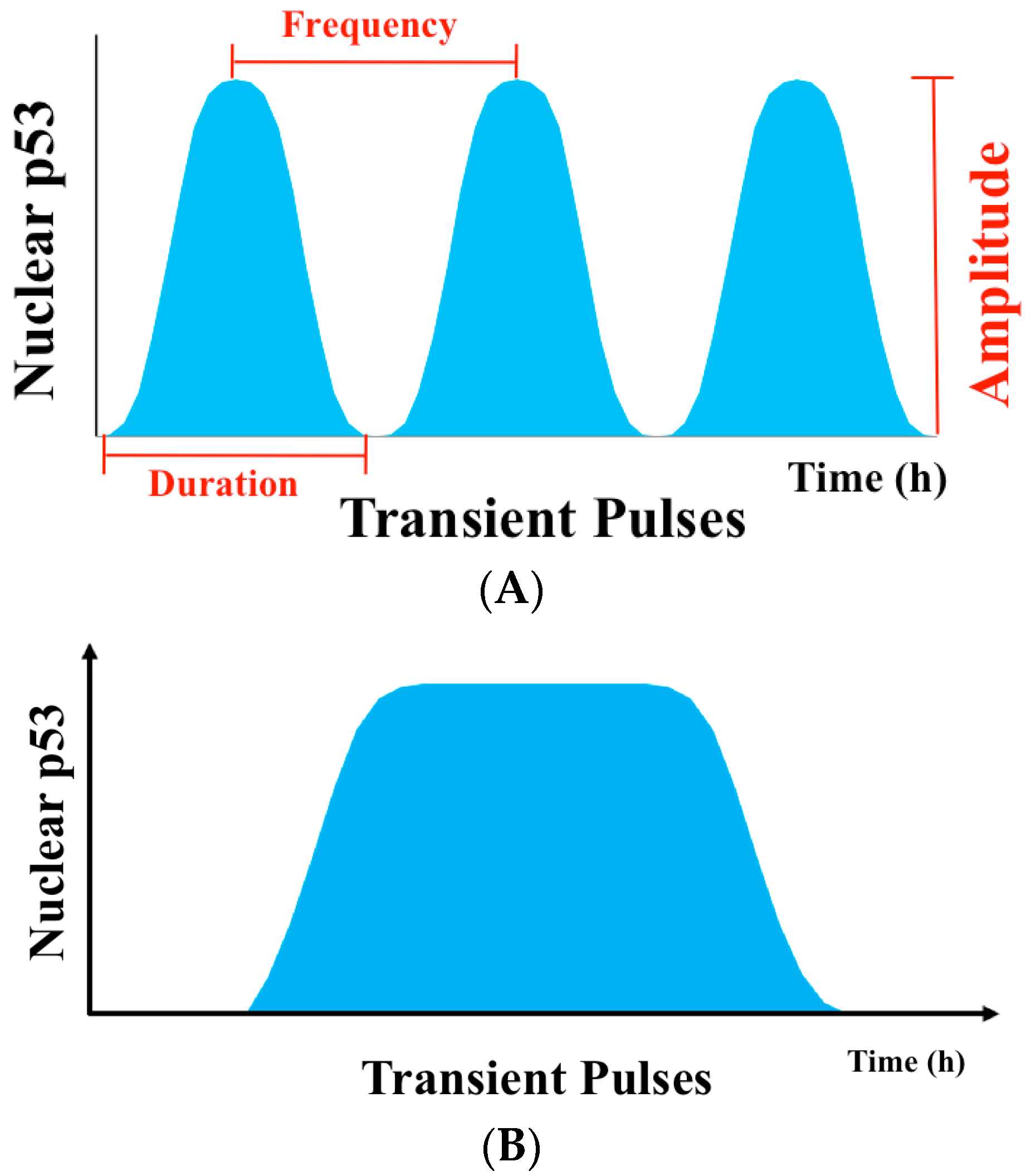
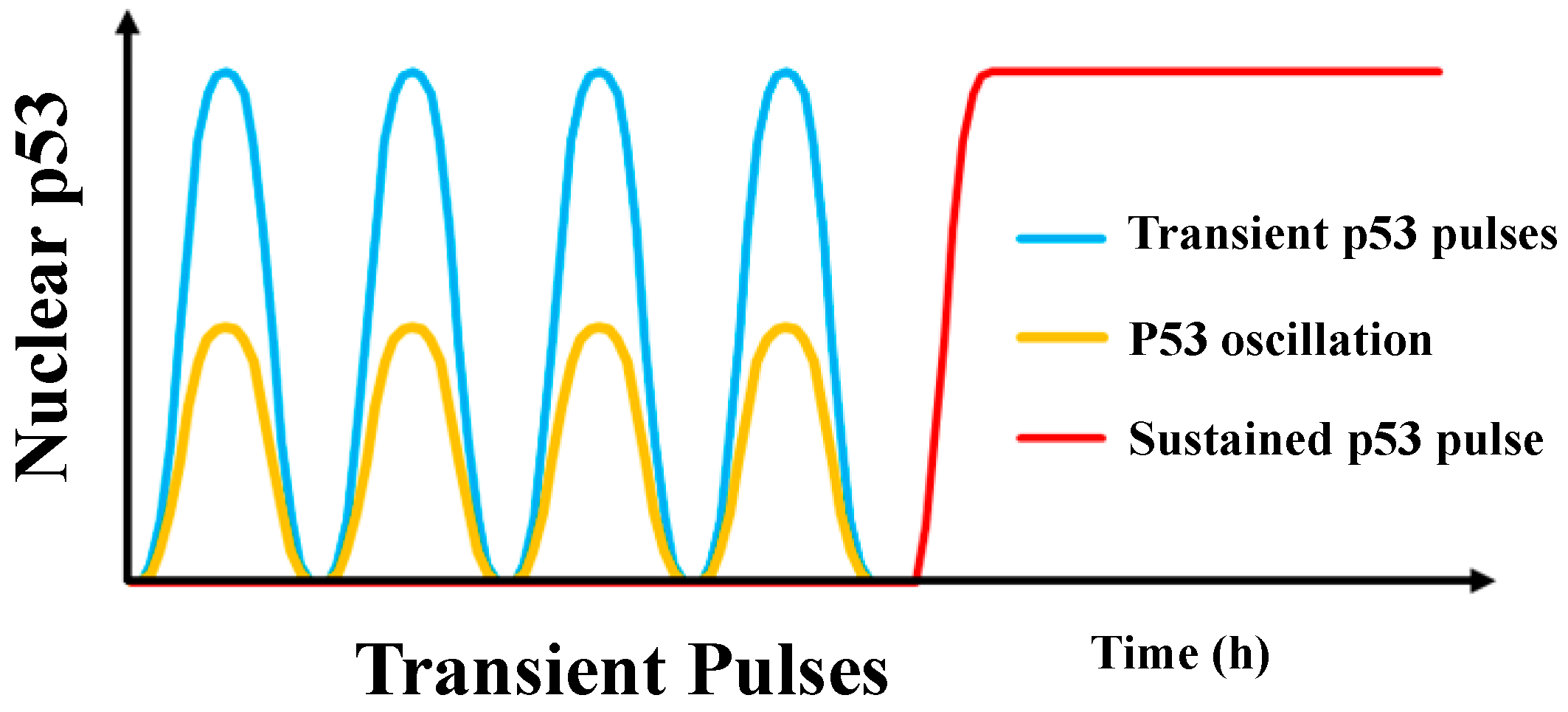
2. Pulsatile p53 Dynamics of Protein Level
2.1. The Dynamics of p53 in Single Cells
2.2. The Role of p53 Dynamics in Making Cell Fate Decisions
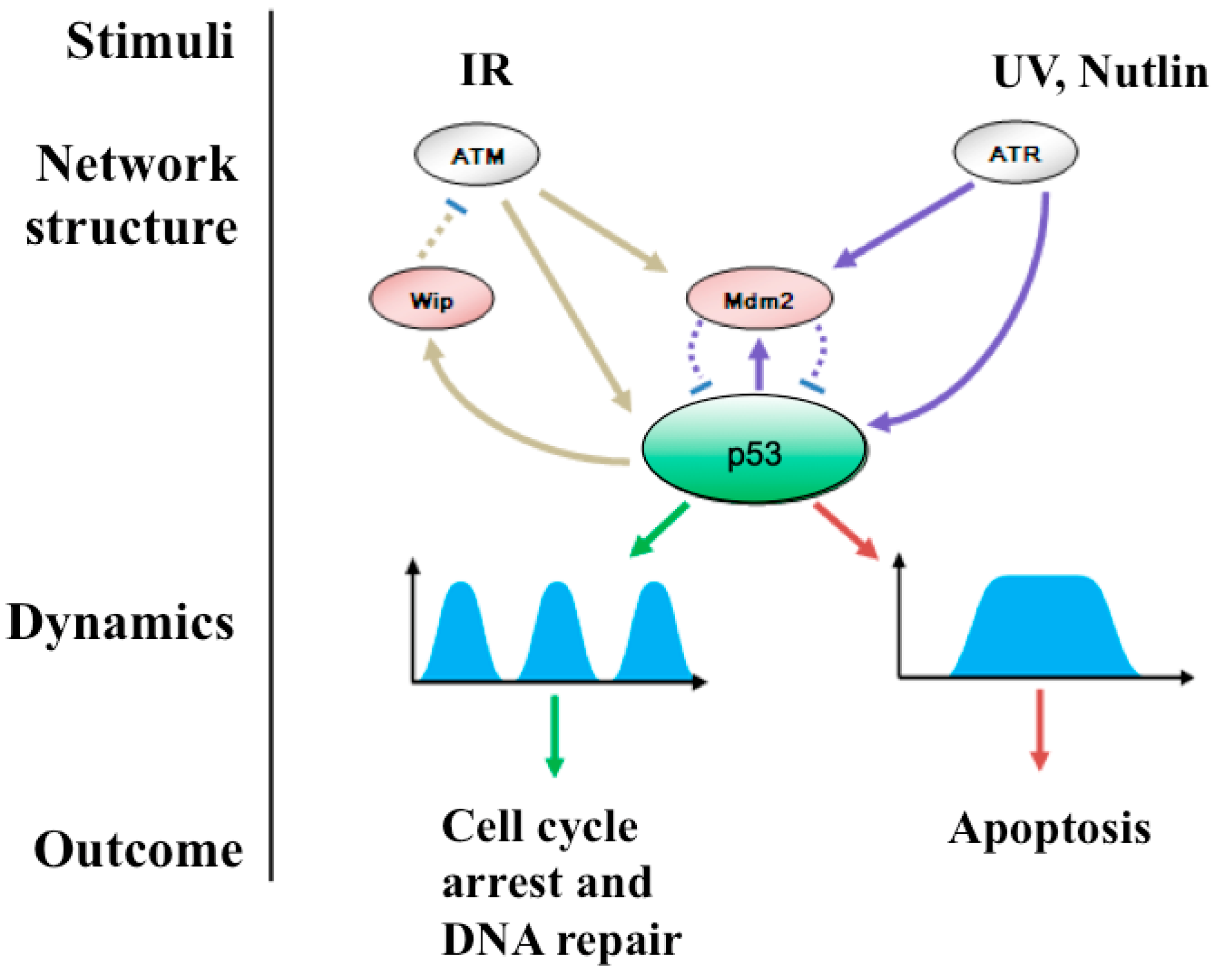
2.2.1. How Can Pulsatile p53 Dynamics Be Generated?
2.2.2. How Does Pulsatile p53 Dynamics Make the Cell Fate Decision?
3. The Dynamics of p53 That Are Independent of Its Cellular Protein Level
3.1. The Spatial Dynamics of p53 in Cells
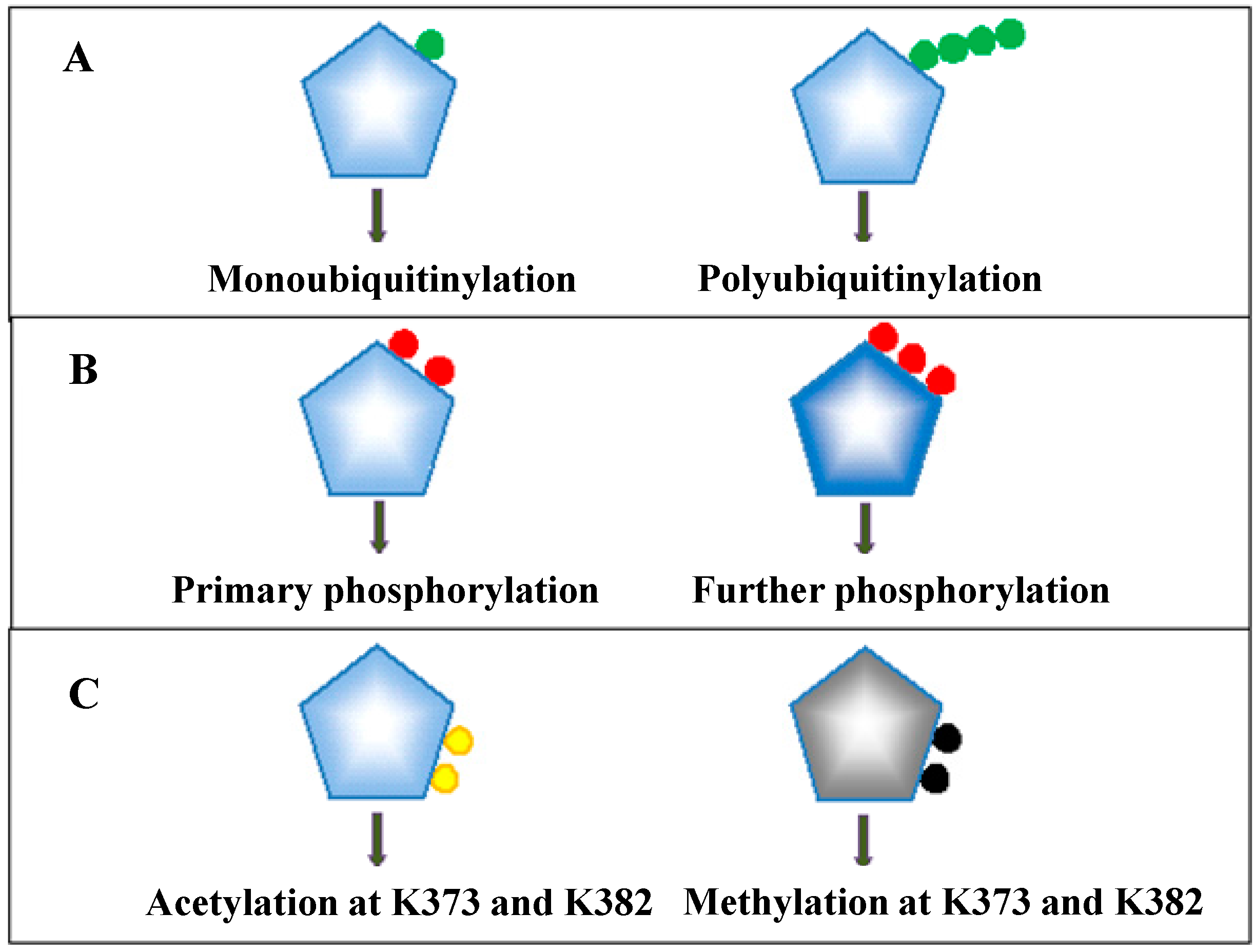
3.2. The Dynamics of Posttranslational Modifications of p53
3.3. Roles of MicroRNAs (miRNAs) in Modulating p53 Dynamics
3.4. The Network of p53 Dynamics in Governing Cell Fate and Cancer Therapy
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [PubMed]
- Wang, P.; Yu, J.; Zhang, L. The nuclear function of p53 is required for puma-mediated apoptosis induced by DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 4054–4059. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. p53 enters the microRNA world. Cancer Cell 2007, 12, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Loewer, A.; Lahav, G. The ups and downs of p53: Understanding protein dynamics in single cells. Nat. Rev. Cancer 2009, 9, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Purvis, J.E.; Lahav, G. Encoding and decoding cellular information through signaling dynamics. Cell 2013, 152, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.J.; Liu, F.; Zhang, X.P.; Li, J.; Wang, W. A two-step mechanism for cell fate decision by coordination of nuclear and mitochondrial p53 activities. PLoS ONE 2012, 7, e38164. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Scimeca, J.C.; Filloux, C.; Peraldi, P.; Carpentier, J.L.; van Obberghen, E. Co-regulation of the mitogen-activated protein kinase, extracellular signal-regulated kinase 1, and the 90-kDa ribosomal s6 kinase in PC12 cells. Distinct effects of the neurotrophic factor, nerve growth factor, and the mitogenic factor, epidermal growth factor. J. Biol. Chem. 1993, 268, 9803–9810. [Google Scholar] [PubMed]
- Xing, J.; Kornhauser, J.M.; Xia, Z.; Thiele, E.A.; Greenberg, M.E. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate creb serine 133 phosphorylation. Mol. Cell. Biol. 1998, 18, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Groot, M.; Boxer, L.M.; Thiel, G. Nerve growth factor- and epidermal growth factor-regulated gene transcription in PC12 pheochromocytoma and INS-1 insulinoma cells. Eur. J. Cell Biol. 2000, 79, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Liu, F.; Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed]
- Lev Bar-Or, R.; Maya, R.; Segel, L.A.; Alon, U.; Levine, A.J.; Oren, M. Generation of oscillations by the p53-Mdm2 feedback loop: A theoretical and experimental study. Proc. Natl. Acad. Sci. USA 2000, 97, 11250–11255. [Google Scholar] [CrossRef] [PubMed]
- Hamstra, D.A.; Bhojani, M.S.; Griffin, L.B.; Laxman, B.; Ross, B.D.; Rehemtulla, A. Real-time evaluation of p53 oscillatory behavior in vivo using bioluminescent imaging. Cancer Res. 2006, 66, 7482–7489. [Google Scholar] [CrossRef] [PubMed]
- Geva-Zatorsky, N.; Rosenfeld, N.; Itzkovitz, S.; Milo, R.; Sigal, A.; Dekel, E.; Yarnitzky, T.; Liron, Y.; Polak, P.; Lahav, G.; et al. Oscillations and variability in the p53 system. Mol. Syst. Biol. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchelor, E.; Loewer, A.; Mock, C.; Lahav, G. Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell 2016, 165, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vousden, K.H. The role of ubiquitin modification in the regulation of p53. Biochim. Biophys. Acta 2014, 1843, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Barak, Y.; Juven, T.; Haffner, R.; Oren, M. Mdm2 expression is induced by wild type p53 activity. EMBO J. 1993, 12, 461–468. [Google Scholar] [PubMed]
- Ye, M.; Tang, Y.; Tang, S.; Liu, J.; Wu, K.; Yao, S.; Sun, Y.; Zhou, L.; Deng, T.; Chen, Y.; et al. Stip is a critical nuclear scaffolding protein linking usp7 to p53-Mdm2 pathway regulation. Oncotarget 2015, 6, 34718–34731. [Google Scholar] [PubMed]
- Meng, X.; Franklin, D.A.; Dong, J.; Zhang, Y. Mdm2-p53 pathway in hepatocellular carcinoma. Cancer Res. 2014, 74, 7161–7167. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Pant, V.; Lozano, G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 2014, 28, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Christov, K.; Shilkaitis, A.; Bratescu, L.; Green, A.; Santini, S.; Bizzarri, A.R.; Cannistraro, S.; Gupta, T.K.; Beattie, C.W. p28, a first in class peptide inhibitor of cop1 binding to p53. Br. J. Cancer 2013, 108, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Nguyen, T.A.; Donehower, L.A. Reversal of the ATM/ATR-mediated DNA damage response by the oncogenic phosphatase PPM1D. Cell Cycle 2005, 4, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent initiation: A mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell 2008, 30, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Liu, F.; Cheng, Z.; Wang, W. Cell fate decision mediated by p53 pulses. Proc. Natl. Acad. Sci. USA 2009, 106, 12245–12250. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jackson, T.L. Mechanisms that enhance sustainability of p53 pulses. PLoS ONE 2013, 8, e65242. [Google Scholar] [CrossRef] [PubMed]
- Loewer, A.; Batchelor, E.; Gaglia, G.; Lahav, G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell 2010, 142, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Lahav, G. The strength of indecisiveness: Oscillatory behavior for better cell fate determination. Sci. Signal. 2004. [Google Scholar] [CrossRef] [PubMed]
- Deguin-Chambon, V.; Vacher, M.; Jullien, M.; May, E.; Bourdon, J.C. Direct transactivation of c-Ha-Ras gene by p53: Evidence for its involvement in p53 transactivation activity and p53-mediated apoptosis. Oncogene 2000, 19, 5831–5841. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Erster, S.; Mihara, M.; Kim, R.H.; Petrenko, O.; Moll, U.M. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol. Cell. Biol. 2004, 24, 6728–6741. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Wolff, S.; Speidel, D.; Deppert, W. Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell Biol. 2005, 17, 631–636. [Google Scholar] [CrossRef] [PubMed]
- David, R. Apoptosis: A lipid trigger of momp. Nat. Rev. Mol. Cell Biol. 2012, 13, 208–209. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Marouco, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. Lysine-specific modifications of p53: A matter of life and death? Oncotarget 2013, 4, 1556–1571. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono-versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Tavana, O.; Gu, W. Modulation of the p53/Mdm2 interplay by HAUSP inhibitors. J. Mol. Cell Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vigneron, A.M.; Vousden, K.H. Ubiquitin-specific peptidase 42 (USP42) functions to deubiquitylate histones and regulate transcriptional activity. J. Biol. Chem. 2014, 289, 34862–34870. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [PubMed]
- Dashzeveg, N.; Taira, N.; Lu, Z.G.; Kimura, J.; Yoshida, K. Palmdelphin, a novel target of p53 with Ser46 phosphorylation, controls cell death in response to DNA damage. Cell Death Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, G.S.; Ivanova, T.; Kurash, J.; Ivanov, A.; Chuikov, S.; Gizatullin, F.; Herrera-Medina, E.M.; Rauscher, F., 3rd; Reinberg, D.; Barlev, N.A. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell. Biol. 2007, 27, 6756–6769. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. Roles for microRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, Q.; Zhang, Z.; Ge, S.; Han, Z.G.; Chen, W.T. Loss of microRNA-143/145 disturbs cellular growth and apoptosis of human epithelial cancers by impairing the Mdm2-p53 feedback loop. Oncogene 2013, 32, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hattori, H.; Janky, R.; Nietfeld, W.; Aerts, S.; Madan Babu, M.; Venkitaraman, A.R. p53 shapes genome-wide and cell type-specific changes in microRNA expression during the human DNA damage response. Cell Cycle 2014, 13, 2572–2586. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M.; Reed, J.C. Finally, an apoptosis-targeting therapeutic for cancer. Cancer Res. 2016, 76, 5914–5920. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.H.; Zhang, X.P.; Liu, F.; Wang, W. Involvement of miR-605 and miR-34a in the DNA damage response promotes apoptosis induction. Biophys. J. 2014, 106, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Lin, H.; Luo, X.; Luo, X.; Wang, Z. miR-605 joins p53 network to form a p53:miR-605:Mdm2 positive feedback loop in response to stress. EMBO J. 2011, 30, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.S.; Wang, Y.; Guo, S.; Zhao, X.; Song, B. Influence of microRNA 34a on proliferation, invasion and metastasis of HCT116 cells. Mol. Med. Rep. 2017, 15, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ludwig, R.L.; Jensen, J.P.; Pierre, S.A.; Medaglia, M.V.; Davydov, I.V.; Safiran, Y.J.; Oberoi, P.; Kenten, J.H.; Phillips, A.C.; et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005, 7, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Reece, K.M.; Figg, W.D. A novel regulator (USP10) of p53: Implications for tumor suppression and therapeutic targeting. Cancer Biol. Ther. 2010, 9, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Dayaram, T.; Gilmartin, A.G.; Ganji, G.; Pemmasani, S.K.; van der Key, H.; Shohet, J.M.; Donehower, L.A.; Kumar, R. Wip1 phosphatase as a potential therapeutic target in neuroblastoma. PLoS ONE 2015, 10, e0115635. [Google Scholar] [CrossRef] [PubMed]
- Demir, O.; Ieong, P.U.; Amaro, R.E. Full-length p53 tetramer bound to DNA and its quaternary dynamics. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Saxena, R.; Sinclair, E.; Fu, Y.; Jacobs, A.; Dyba, M.; Wang, X.; Cruz, I.; Berry, D.; Kallakury, B.; et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016, 23, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Sales, F.; van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.C.; et al. p53 reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K)braf melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Q.; Beaver, J.M.; Liu, Y.; Zhang, Z. Dynamics of p53: A Master Decider of Cell Fate. Genes 2017, 8, 66. https://doi.org/10.3390/genes8020066
Luo Q, Beaver JM, Liu Y, Zhang Z. Dynamics of p53: A Master Decider of Cell Fate. Genes. 2017; 8(2):66. https://doi.org/10.3390/genes8020066
Chicago/Turabian StyleLuo, Qingyin, Jill M. Beaver, Yuan Liu, and Zunzhen Zhang. 2017. "Dynamics of p53: A Master Decider of Cell Fate" Genes 8, no. 2: 66. https://doi.org/10.3390/genes8020066
APA StyleLuo, Q., Beaver, J. M., Liu, Y., & Zhang, Z. (2017). Dynamics of p53: A Master Decider of Cell Fate. Genes, 8(2), 66. https://doi.org/10.3390/genes8020066