
The Ageing Brain: Effects on DNA Repair and DNA Methylation in Mice


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Design of the Study
2.2. Determination of 8-oxodG
2.3. Assessment of Genomic 5mC and 5hmC
2.4. Gene-Specific Methylation Studies, Using Pyrosequencing of Bisulphite Converted DNA
2.5. Gene Expression Analyses
2.6. Measurement of BER-Related DNA Incision Activity
2.7. Statistical Analysis
3. Results
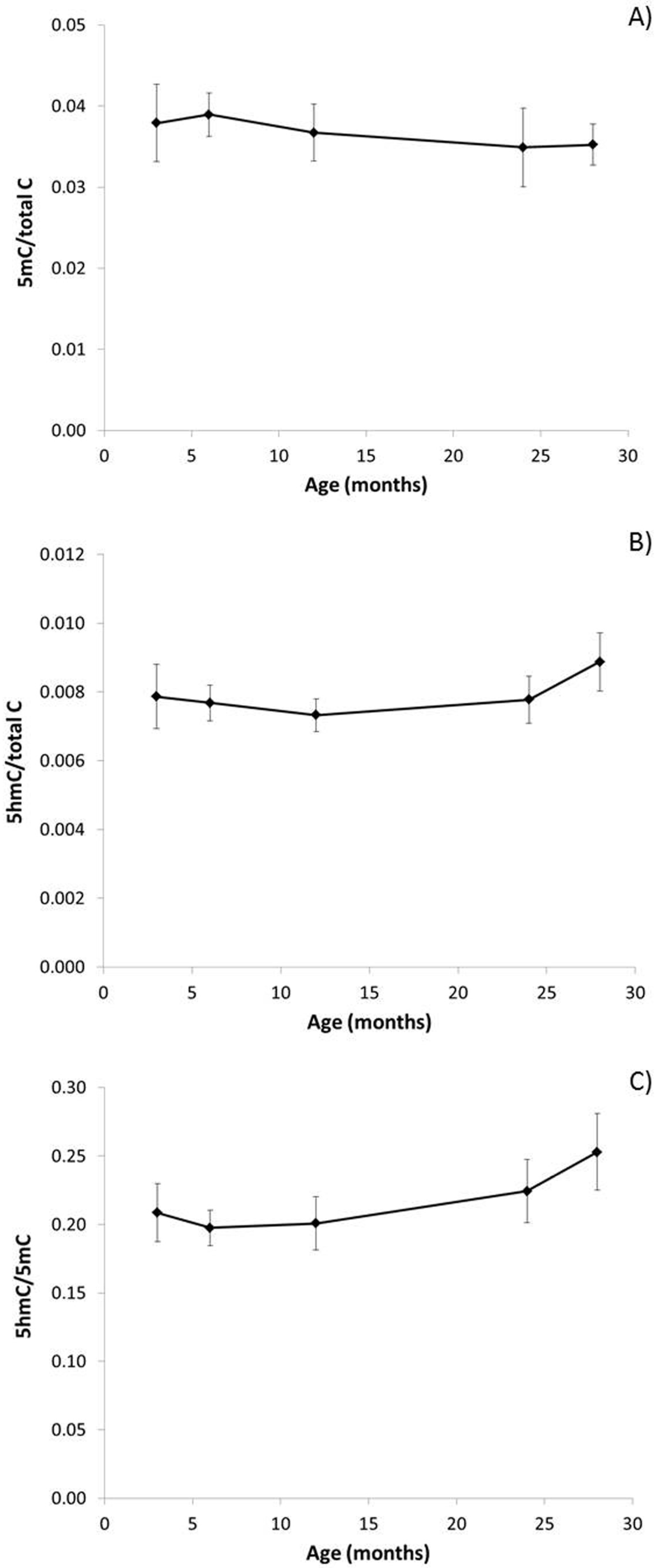
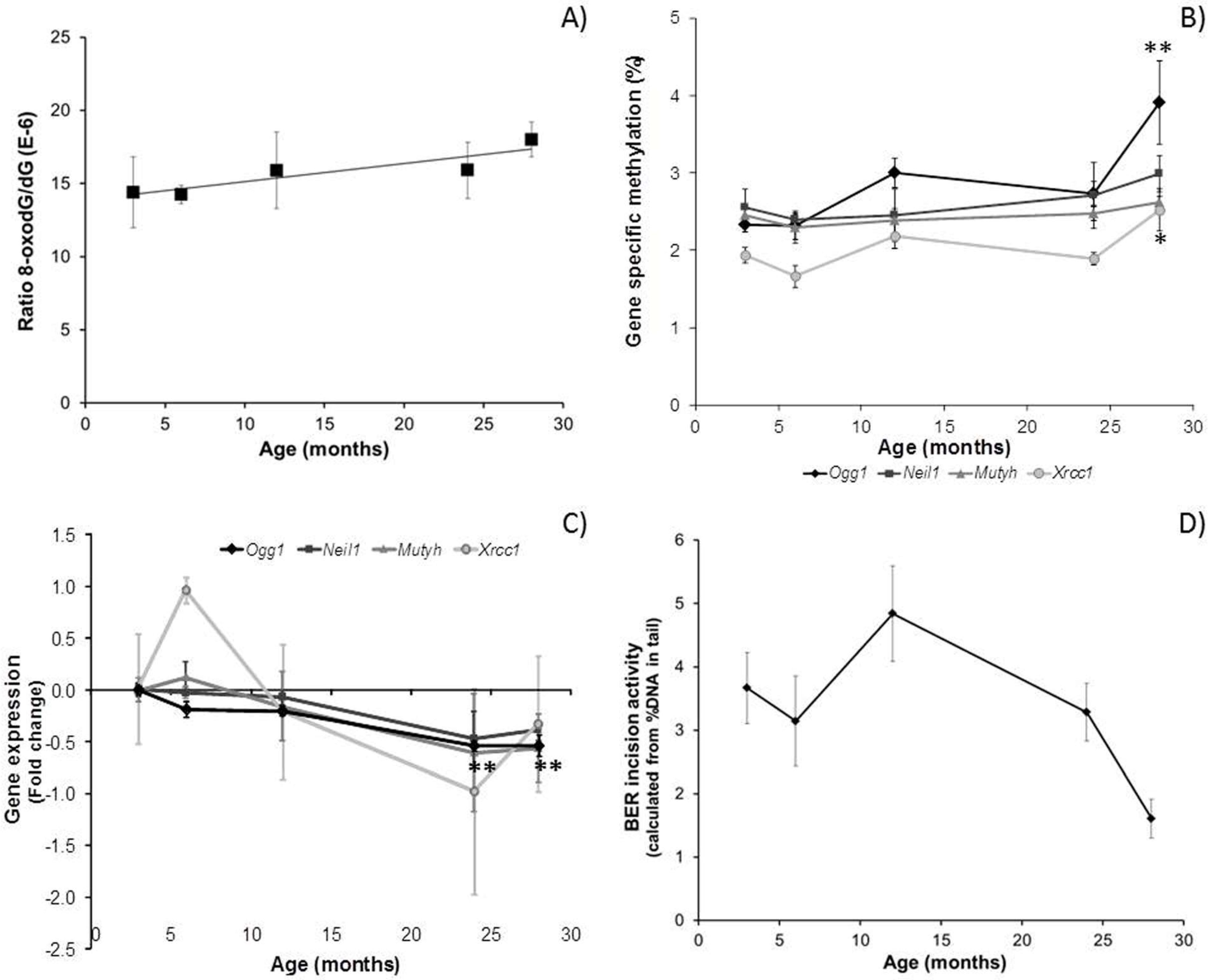
3.1. The Effect of Ageing on Genome Stability, DNA Damage and DNA Methylation
3.2. Effect of Ageing on Gene Expression in Mouse Brain
3.3. Phenotypic Effects in the Ageing Brain
3.4. Involvement of Tet Enzymes and Methyl-CpG Binding Proteins
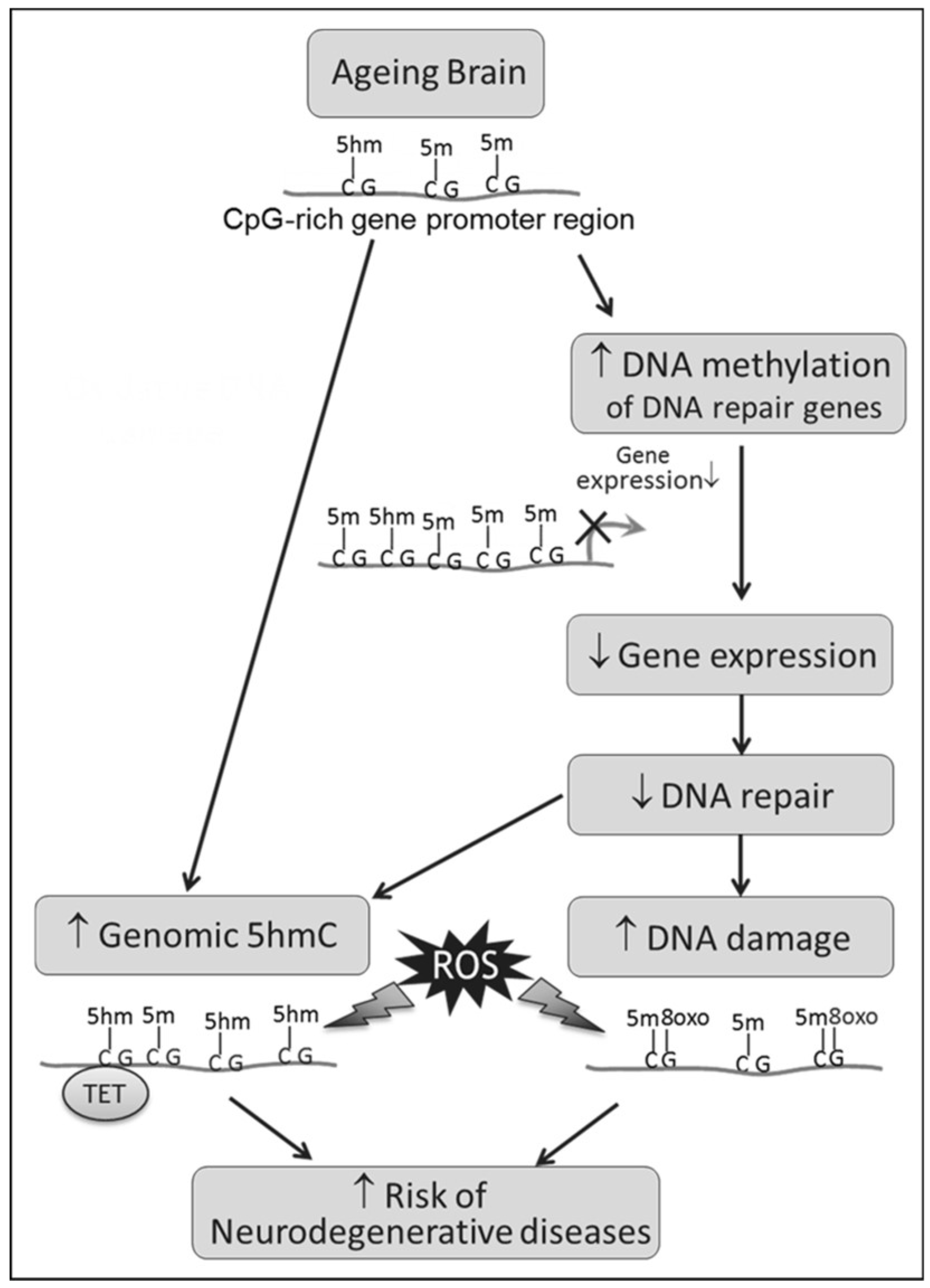
4. Discussion
4.1. Age-Related Increase in 5hmC, a Result of TET2 or Decreased BER?
4.2. Epigenetic Regulation of Ogg1 Plays a Role in Age-Related Decline in DNA Repair
4.3. Concluding Remarks
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 5caC | 5-carboxylcytosine |
| 5fC | 5-formylcytosine |
| 5hmC | 5-hydroxymethycytosine |
| 5mC | 5-methylcytosine |
| 8-oxodG | 8-oxo-7,8-dihydro-2′-deoxyguanosine |
| BER | base excision repair |
| Mecp2 | methyl-CpG binding protein 2 |
| Mutyh | mutY DNA glycosylase |
| nDNA | nuclear DNA |
| NER | nucleotide excision repair |
| Neil1 | Nei-like DNA glycosylase 1 |
| mtDNA | mitochondrial DNA |
| Ogg1 | DNA glycosylase oxoguanosine 1 |
| ROS | reactive oxygen species; |
| TDG | thymine DNA glycosylase |
| TEMPO | 2,2,6,6-tetramethylpiperidine-N-oxyl |
| TET | ten-eleven translocation enzymes |
| TF | transcription factors |
| Xrcc1 | X-ray repair cross-complementing protein 1 |
References
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Langie, S.A.S.; Lara, J.; Mathers, J.C. Early determinants of the ageing trajectory. Best Pract. Res. Clin. Endocrinol. Metab. 2012, in press. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Zharkov, D.O.; Dianov, G.L. NEIL1 excises 3′ end proximal oxidative DNA lesions resistant to cleavage by NTH1 and OGG1. Nucleic Acids Res. 2005, 33, 4849–4856. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef]
- Marsin, S.; Vidal, A.E.; Sossou, M.; Menissier-de Murcia, J.; Le Page, F.; Boiteux, S.; de Murcia, G.; Radicella, J.P. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J. Biol. Chem. 2003, 278, 44068–44074. [Google Scholar] [CrossRef] [PubMed]
- Osterod, M.; Hollenbach, S.; Hengstler, J.G.; Barnes, D.E.; Lindahl, T.; Epe, B. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis 2001, 22, 1459–1463. [Google Scholar] [CrossRef]
- Chen, S.K.; Hsieh, W.A.; Tsai, M.H.; Chen, C.C.; Hong, A.I.; Wei, Y.H.; Chang, W.P. Age-associated decrease of oxidative repair enzymes, human 8-oxoguanine DNA glycosylases (hOgg1), in human aging. J. Radiat. Res. 2003, 44, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Jacob, K.D.; Noren Hooten, N.; Tadokoro, T.; Lohani, A.; Barnes, J.; Evans, M.K. Alzheimer’s disease-associated polymorphisms in human OGG1 alter catalytic activity and sensitize cells to DNA damage. Free Radic. Biol. Med. 2013, 63, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Pan, X.; Zhu, B.B.; Zhang, Y.; Yuan, F.; Huang, J.; Lovell, M.A.; Lee, M.P.; Markesbery, W.R.; Li, G.M.; et al. Identification and characterization of OGG1 mutations in patients with alzheimer’s disease. Nucleic Acids Res. 2007, 35, 2759–2766. [Google Scholar] [CrossRef] [PubMed]
- Cabelof, D.C.; Raffoul, J.J.; Yanamadala, S.; Ganir, C.; Guo, Z.; Heydari, A.R. Attenuation of DNA polymerase beta-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat. Res. 2002, 500, 135–145. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Fitzpatrick, M.; Kompaniez, K.; Jacob, K.D.; Moore, B.R.; Nagle, J.; Barnes, J.; Lohani, A.; Evans, M.K. Coordination of DNA repair by NEIL1 and PARP-1: A possible link to aging. Aging 2012, 4, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar] [PubMed]
- Langie, S.A.; Koppen, G.; Desaulniers, D.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Azqueta, A.; Bisson, W.H.; Brown, D.G.; Brunborg, G.; et al. Causes of genome instability: The effect of low dose chemical exposures in modern society. Carcinogenesis 2015, 36 (Suppl. S1), S61–S88. [Google Scholar] [CrossRef] [PubMed]
- Langie, S.A.; Kowalczyk, P.; Tomaszewski, B.; Vasilaki, A.; Maas, L.M.; Moonen, E.J.; Palagani, A.; Godschalk, R.W.; Tudek, B.; van Schooten, F.J.; et al. Redox and epigenetic regulation of the APE1 gene in the hippocampus of piglets: The effect of early life exposures. DNA Repair 2014, 18, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Mehler, M.F. Epigenetic principles and mechanisms underlying nervous system functions in health and disease. Prog. Neurobiol. 2008, 86, 305–341. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Calvanese, V.; Lara, E.; Kahn, A.; Fraga, M.F. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009, 8, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Mathers, J.C.; Strathdee, G.; Relton, C.L. Induction of epigenetic alterations by dietary and other environmental factors. In Advances in Genetics; Herceg, Z., Ushijima, T., Eds.; Academic Press: Burlington, MA, USA, 2010; Volume 71, pp. 1–39. [Google Scholar]
- Nabel, C.S.; Kohli, R.M. Molecular biology. Demystifying DNA demethylation. Science 2011, 333, 1229–1230. [Google Scholar] [CrossRef] [PubMed]
- Van den Hove, D.L.; Chouliaras, L.; Rutten, B.P. The role of 5-hydroxymethylcytosine in aging and Alzheimer’s disease: Current status and prospects for future studies. Curr. Alzheimer Res. 2012, 9, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Kasahara, I.; Sawabe, M.; Honma, N.; Aida, J.; Tabubo, K. Role of methylation of the hMLH1 gene promoter in the development of gastric and colorectal carcinoma in the elderly. Geriatr. Gerontol. Int. 2010, 10 (Suppl. S1), S207–S212. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.M. Epigenetics, mismatch repair genes and colorectal cancer. Ann. R. Coll. Surg. Engl. 2005, 87, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Agrelo, R.; Cheng, W.H.; Setien, F.; Ropero, S.; Espada, J.; Fraga, M.F.; Herranz, M.; Paz, M.F.; Sanchez-Cespedes, M.; Artiga, M.J.; et al. Epigenetic inactivation of the premature aging werner syndrome gene in human cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 8822–8827. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. DNA repair deficiency and neurological disease. Nat. Rev. Neurosci. 2009, 10, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T.; Hanawalt, P.C. When parsimony backfires: Neglecting DNA repair may doom neurons in alzheimer’s disease. BioEssays 2003, 25, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Rowlatt, C.; Chesterman, F.C.; Sheriff, M.U. Lifespan, age changes and tumour incidence in an ageing C57BL mouse colony. Lab. Anim. 1976, 10, 419–442. [Google Scholar] [CrossRef] [PubMed]
- Godschalk, R.W.; Maas, L.M.; van Zandwijk, N.; van’t Veer, L.J.; Breedijk, A.; Borm, P.J.; Verhaert, J.; Kleinjans, J.C.; van Schooten, F.J. Differences in aromatic-DNA adduct levels between alveolar macrophages and subpopulations of white blood cells from smokers. Carcinogenesis 1998, 19, 819–825. [Google Scholar] [CrossRef] [PubMed]
- European Standards Committee on Oxidative DNA Damage. Comparison of different methods of measuring 8-oxoguanine as a marker of oxidative DNA damage. Free Radic. Res. 2000, 32, 333–341. [Google Scholar]
- Langie, S.A.; Kowalczyk, P.; Tudek, B.; Zabielski, R.; Dziaman, T.; Olinski, R.; van Schooten, F.J.; Godschalk, R.W. The effect of oxidative stress on nucleotide-excision repair in colon tissue of newborn piglets. Mutat. Res. 2010, 695, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Leung, F.C. An evaluation of new criteria for cpg islands in the human genome as gene markers. Bioinformatics 2004, 20, 1170–1177. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Langie, S.A.; Cameron, K.M.; Waldron, K.J.; Fletcher, K.P.; von Zglinicki, T.; Mathers, J.C. Measuring DNA repair incision activity of mouse tissue extracts towards singlet oxygen-induced DNA damage: A comet-based in vitro repair assay. Mutagenesis 2011, 26, 461–471. [Google Scholar] [CrossRef]
- Borgesius, N.Z.; de Waard, M.C.; van der Pluijm, I.; Omrani, A.; Zondag, G.C.; van der Horst, G.T.; Melton, D.W.; Hoeijmakers, J.H.; Jaarsma, D.; Elgersma, Y. Accelerated age-related cognitive decline and neurodegeneration, caused by deficient DNA repair. J. Neurosci. 2011, 31, 12543–12553. [Google Scholar] [CrossRef] [PubMed]
- De Waard, M.C.; van der Pluijm, I.; Zuiderveen Borgesius, N.; Comley, L.H.; Haasdijk, E.D.; Rijksen, Y.; Ridwan, Y.; Zondag, G.; Hoeijmakers, J.H.; Elgersma, Y.; et al. Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol. 2010, 120, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Mathers, J.C.; Coxhead, J.M.; Tyson, J. Nutrition and DNA repair—Potential molecular mechanisms of action. Curr. Cancer Drug Targets 2007, 7, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Zawia, N.H.; Lahiri, D.K.; Cardozo-Pelaez, F. Epigenetics, oxidative stress, and alzheimer disease. Free Radic. Biol. Med. 2009, 46, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.C.; Boileau, P.; Le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child health, developmental plasticity, and epigenetic programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Steinbacher, J.; Kraus, T.F.; Michalakis, S.; Hackner, B.; Pfaffeneder, T.; Perera, A.; Muller, M.; Giese, A.; Kretzschmar, H.A.; et al. Age-dependent levels of 5-methyl-, 5-hydroxymethyl-, and 5-formylcytosine in human and mouse brain tissues. Angew. Chem. Int. Ed. Engl. 2015, 54, 12511–12514. [Google Scholar] [CrossRef] [PubMed]
- Madrid, A.; Papale, L.A.; Alisch, R.S. New hope: The emerging role of 5-hydroxymethylcytosine in mental health and disease. Epigenomics 2016, 8, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Munzel, M.; Globisch, D.; Carell, T. 5-hydroxymethylcytosine, the sixth base of the genome. Angew. Chem. Int. Ed. Engl. 2011, 50, 6460–6468. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Shi, Y.G. TET family proteins and 5-hydroxymethylcytosine in development and disease. Development 2012, 139, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.; Antunes, C.; Guedes, M.; Sousa, N.; Marques, C.J. TET enzymes and DNA hydroxymethylation in neural development and function—How critical are they? Genomics 2014, 104, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, D.J.; Liu, M.Y.; Nabel, C.S.; Cao, X.J.; Garcia, B.A.; Kohli, R.M. Tet2 catalyzes stepwise 5-methylcytosine oxidation by an iterative and de novo mechanism. J. Am. Chem. Soc. 2016, 138, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Mahfoudhi, E.; Talhaoui, I.; Cabagnols, X.; Della Valle, V.; Secardin, L.; Rameau, P.; Bernard, O.A.; Ishchenko, A.A.; Abbes, S.; Vainchenker, W.; et al. Tet2-mediated 5-hydroxymethylcytosine induces genetic instability and mutagenesis. DNA Repair 2016, 43, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Branco, M.R.; Ficz, G.; Oxley, D.; Krueger, F.; Reik, W.; Balasubramanian, S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.R.; Ficz, G.; Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nature Rev. Genet. 2012, 13, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Izumi, H.; Uchiumi, T.; Suzuki, H.; Arao, T.; Nishio, K.; Kohno, K. ZNF143 interacts with p73 and is involved in cisplatin resistance through the transcriptional regulation of DNA repair genes. Oncogene 2007, 26, 5194–5203. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, A.; Aruga, J. Functional role of Zic2 phosphorylation in transcriptional regulation. FEBS Lett. 2008, 582, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A. Dynamic epigenetic regulation in neurons: Enzymes, stimuli and signaling pathways. Nat. Neurosci. 2010, 13, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Gorniak, J.; Langie, S.A.S.; Cameron, K.; von Zglinicki, T.; Mathers, J.C. The effect of ageing and short-term dietary restriction on the epigenetic, transcriptomic and phenotypic profile of base excision repair in mouse brain and liver. Proc. Nutr. Soc. 2012, 71, E56. [Google Scholar] [CrossRef]
- Cabelof, D.C.; Yanamadala, S.; Raffoul, J.J.; Guo, Z.; Soofi, A.; Heydari, A.R. Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline. DNA Repair 2003, 2, 295–307. [Google Scholar] [CrossRef]
- Imam, S.Z.; Karahalil, B.; Hogue, B.A.; Souza-Pinto, N.C.; Bohr, V.A. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol. Aging 2006, 27, 1129–1136. [Google Scholar] [CrossRef]
- Xu, G.; Herzig, M.; Rotrekl, V.; Walter, C.A. Base excision repair, aging and health span. Mech. Ageing Dev. 2008, 129, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.S. Dietary calorie restriction, DNA-repair and brain aging. Mol. Cell. Biochem. 2003, 253, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Gedik, C.M.; Grant, G.; Morrice, P.C.; Wood, S.G.; Collins, A.R. Effects of age and dietary restriction on oxidative DNA damage, antioxidant protection and DNA repair in rats. Eur. J. Nutr. 2005, 44, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Lohr, M.; Folkmann, J.K.; Mikkelsen, L.; Loft, S. Aging and oxidatively damaged nuclear DNA in animal organs. Free Radic. Biol. Med. 2010, 48, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Langie, S.A.; Achterfeldt, S.; Gorniak, J.P.; Halley-Hogg, K.J.; Oxley, D.; van Schooten, F.J.; Godschalk, R.W.; McKay, J.A.; Mathers, J.C. Maternal folate depletion and high-fat feeding from weaning affects DNA methylation and DNA repair in brain of adult offspring. FASEB J. 2013, 27, 3323–3334. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Queiros, A.C.; Beekman, R.; Martin-Subero, J.I. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim. Biophys. Acta 2013, 1829, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Doi, A.; Park, I.H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific cpg island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef]
- Hon, G.C.; Hawkins, R.D.; Caballero, O.L.; Lo, C.; Lister, R.; Pelizzola, M.; Valsesia, A.; Ye, Z.; Kuan, S.; Edsall, L.E.; et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012, 22, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.; van Dijk, C.M.; Tollenaar, R.A.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011, 44, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Gredilla, R.; Bohr, V.A.; Stevnsner, T. Mitochondrial DNA repair and association with aging—An update. Exp. Gerontol. 2010, 45, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered mitochondrial DNA methylation pattern in alzheimer disease-related pathology and in parkinson disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.M.; Barrow, T.M. Analysis of pollutant-induced changes in mitochondrial DNA methylation. Methods Mol. Biol. 2015, 1265, 271–283. [Google Scholar] [PubMed]
- Castegna, A.; Iacobazzi, V.; Infantino, V. The mitochondrial side of epigenetics. Physiol. Genom. 2015, 47, 299–307. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Amplicon | PCR Primers | Annealing temperature (°C) | Product (bp) | Sequencing primers | Sequence to run on pyrosequencer | Length (bp) |
|---|---|---|---|---|---|---|---|
| Ogg1 | 1 | Fw: 5′-GGTTTATTTTTTGAGATAGA-3′ | 43 | 134 | 5′-TTTAGTTAAGTTTTAAA-3′ | C/TGTGTTTTTC/TGTTTTTGTTTATC/TGAGTTTTGGGAC/TGATC/ TGGTGTGTATTATTAC/TGTTTC/TG | 60 |
| Rev: 5′-BIO-ACTAAAACCACATCATTA-3′ | |||||||
| 2 | Fw: 5′-GTAGGTTTTGAGATTGTAT-3′ | 43 | 184 | 5′-GAAAGTTTTGAAATGGTAGA-3′ | GTG/TGGGTTTTTGGTAGTTAATG/TGTTAAGTAGC/TGAGGTTAGTAGGTT AATC/TGTTTTTATTTTATAGGTTC/TGTTATTTC/TG | 79 | |
| Rev: 5′-BIO-ATTTAACCCTAAAAATAAC-3′ | |||||||
| Neil1 | 1 | Fw: 5′-TGAGGTAGTAGTTAGTAAGG-3′ | 52 | 220 | 5′-GTAGTTAGTAAGGGGTTAAT-3′ | TTTAGTAGTTTGTC/TGAATTTTAGAGTAC/TGTTGGG | 34 |
| Rev: 5′-BIO-ACTCTACTCACAATTCTTT-3′ | 5′-GAATGGAGTTTTTTATTTATGA-3′ | GAATTTC/TGGGTGTTGGGTAACTTTTGGACTAGTC/TGC/ TGTAATTC/TGGAGGTGAC/TGAA | 55 | ||||
| 2 | Fw: 5′-AGAATTGTGAGTAGAGTTTTGT-3′ | 52 | 186 | 5′-GTTTTAGTTATTTTAGATTATA-3′ | C/TGTTAGTAGTC/TGGAAAC/TGGC/TGTTGTGTAGAGTTATAAG TAGTTGTATGC/TGAGG | 53 | |
| Rev: 5′-BIO-ATCTTAAATCCCCAAAAATTA-3′ | |||||||
| Mutyh | 1 | Fw: 5′-GGATGGTTATAGAAGTTTAAG-3′ | 46.6 | 164 | 5′-GTTTTAGTTATTTTAGATTATA-3′ | ATTTTTAGTGTGTAGC/TGC/TGTGTAATTGTAAAATTC/TG | 36 |
| Rev: 5′-BIO-TCACTACTCCACTCTACAA-3′ | |||||||
| Xrcc1 | 1 | Fw: 5′-AGGTTTTAGGAAATTTTTAGTT-3′ | 50 | 228 | 5′-TTTAATGATTAGGGTAAA-3′ | TTATAC/TGTAGGATTTAATTATTGAGGTC/TGTTTTTGTTGT TAGGTTTT AGGAGTC/TGAGTTTTTAG/TG | 67 |
| Rev: 5′-BIO-CCCTTAACAACAAACATTC-3′ | |||||||
| 2 | Fw: 5′-TGTTTGTTGTTAAGGGAATT-3′ | 50 | 328 | 5′-GGAGAGGTTTAATYGAGTAT-3′ | GC/TGTAGTGTTGAC/TGTGTGC/TGTC/TGGC/TGC/TGTC/TGC/TGGTT TGAAAGGTTC/TGAGTTTTGC/TGC/TGTTTGC/TGT | 65 | |
| Rev: 5′-BIO-CTCAAAAAACCCCTATCT-3′ | 5′-GGGGTTTTTTYGGAGTTGTAA-3′ | TTTTTTTTTTTTTATTTTTTTGGAC/TGGTC/TGGGC/TGTTTAC/TGGGC/TG TGGATATGTC/TGGAGATTAGTTTTC/TGTTAC/TGTC/TGT | 76 |
| Primer set 1 | Primer set 2 | ||
|---|---|---|---|
| Gene | q-PCR primers | Gene | q-PCR primers |
| Ogg1 | Fw: 5′-TGGCTTCCCAAACCTCCAT-3′ | Mecp2 | Fw: 5′-GAGGAGGCGAGGAGGAGAGA-3′ |
| Rev: 5′-GGCCCAACTTCCTCAGGTG-3′ | Rev: 5′-AACTTCAGTGGCTTGTCTCTGAGG-3′ | ||
| Neil1 | Fw: 5′-GACCCTGAGCCAGAAGATCAG-3′ | Tet1 | Fw: 5′-CCATTCTCACAAGGACATTCACA-3′ |
| Rev: 5′-AGCTGTGTCTCCTGTGACTT-3′ | Rev: 5′-GCAGGACGTGGAGTTGTTCA-3′ | ||
| Mutyh | Fw: 5′-CTGTCTCCCCATATCATCTCTT-3′ | Tet2 | Fw: 5′-GCCATTCTCAGGAGTCACTGC-3′ |
| Rev: 5′-TCACGCTTCTCTTGGTCATAC-3′ | Rev: 5′-ACTTCTCGATTGTCTTCTCTATTGAGG-3′ | ||
| Xrrc1 | Fw: 5′-CTTCTCAAGGCGGACACTTA-3′ | Tet3 | Fw: 5′-GGTCACAGCCTGCATGGACT-3′ |
| Rev: 5′-ATCTGCTCCTCCTTCTCCAA-3′ | Rev: 5′-AGCGATTGTCTTCCTTGGTCAG-3′: | ||
| B2m | Fw: 5′-ATGCTGAAGAACGGGAAAAAAA-3′ | Atp5b | Fw: 5′-GGCCAAGATGTCCTGCTGTT-3′ |
| Rev: 5′-CAGTGTGAGCCAGGATATAGAA-3′ | Rev: 5′-AACTTTGGCATTGTGGAAGG-3′ | ||
| Hprt | Fw: 5′-AGGAGAGAAAGATGTGATTGATATT-3′ | Gapdh | Fw: 5′-AACTTTGGCATTGTGGAAGG-3′ |
| Rev: 5′-TCCACTGAGCAAAACCTCTT-3′ | Rev: 5′-ATGCAGGGATGATGTTCTGG-3′ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langie, S.A.S.; Cameron, K.M.; Ficz, G.; Oxley, D.; Tomaszewski, B.; Gorniak, J.P.; Maas, L.M.; Godschalk, R.W.L.; Van Schooten, F.J.; Reik, W.; et al. The Ageing Brain: Effects on DNA Repair and DNA Methylation in Mice. Genes 2017, 8, 75. https://doi.org/10.3390/genes8020075
Langie SAS, Cameron KM, Ficz G, Oxley D, Tomaszewski B, Gorniak JP, Maas LM, Godschalk RWL, Van Schooten FJ, Reik W, et al. The Ageing Brain: Effects on DNA Repair and DNA Methylation in Mice. Genes. 2017; 8(2):75. https://doi.org/10.3390/genes8020075
Chicago/Turabian StyleLangie, Sabine A. S., Kerry M. Cameron, Gabriella Ficz, David Oxley, Bartłomiej Tomaszewski, Joanna P. Gorniak, Lou M. Maas, Roger W. L. Godschalk, Frederik J. Van Schooten, Wolf Reik, and et al. 2017. "The Ageing Brain: Effects on DNA Repair and DNA Methylation in Mice" Genes 8, no. 2: 75. https://doi.org/10.3390/genes8020075
APA StyleLangie, S. A. S., Cameron, K. M., Ficz, G., Oxley, D., Tomaszewski, B., Gorniak, J. P., Maas, L. M., Godschalk, R. W. L., Van Schooten, F. J., Reik, W., Von Zglinicki, T., & Mathers, J. C. (2017). The Ageing Brain: Effects on DNA Repair and DNA Methylation in Mice. Genes, 8(2), 75. https://doi.org/10.3390/genes8020075