
Immune-Mediated Therapies for Liver Cancer



Abstract
:1. Introduction
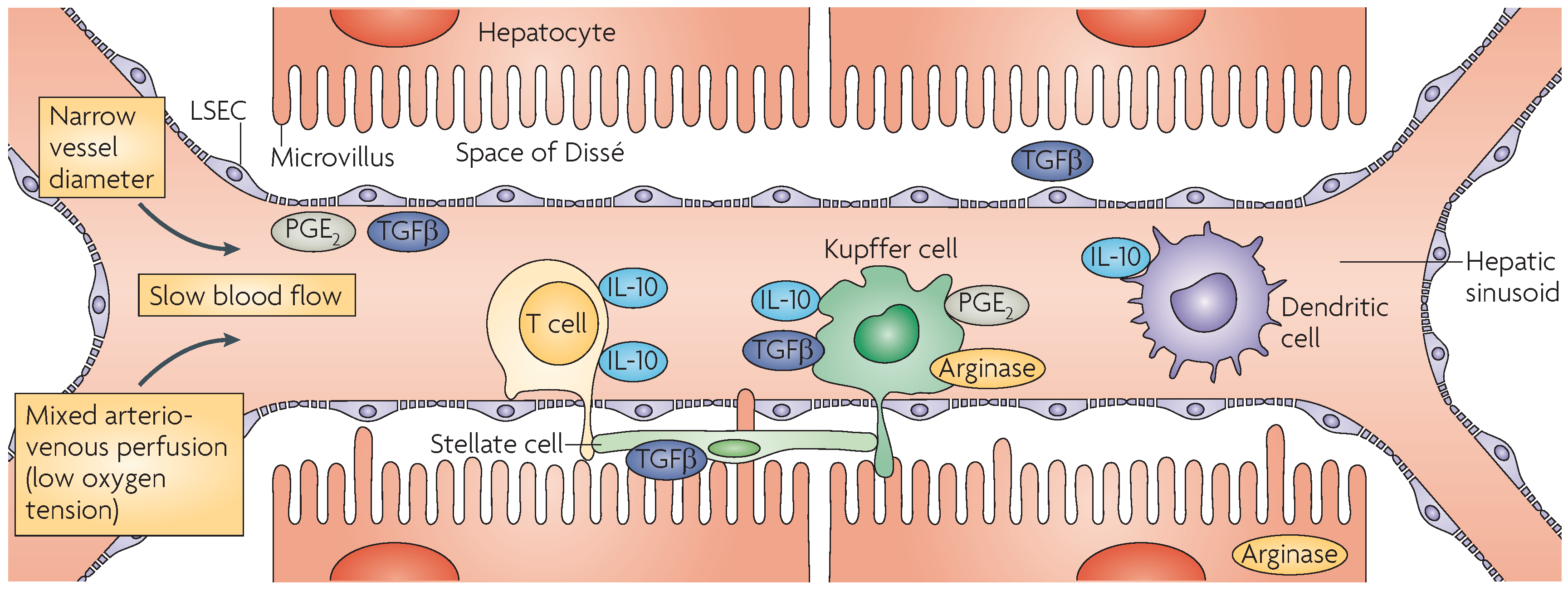
2. Immunobiology of the Liver
2.1. Hepatocytes
2.2. Cholangiocytes
2.3. Kupffer Cells
2.4. Dendritic Cells
2.5. Liver Sinusoidal Endothelial Cells
2.6. Hepatic Stellate Cells
3. Tumor Microenvironment
3.1. Tumor-Infiltrating Lymphocytes
3.2. Tumor-Associated Macrophages
3.3. Innate Immunity in the Liver
4. Immunotherapeutic Approaches to Treat Liver Cancer
4.1. Vaccines
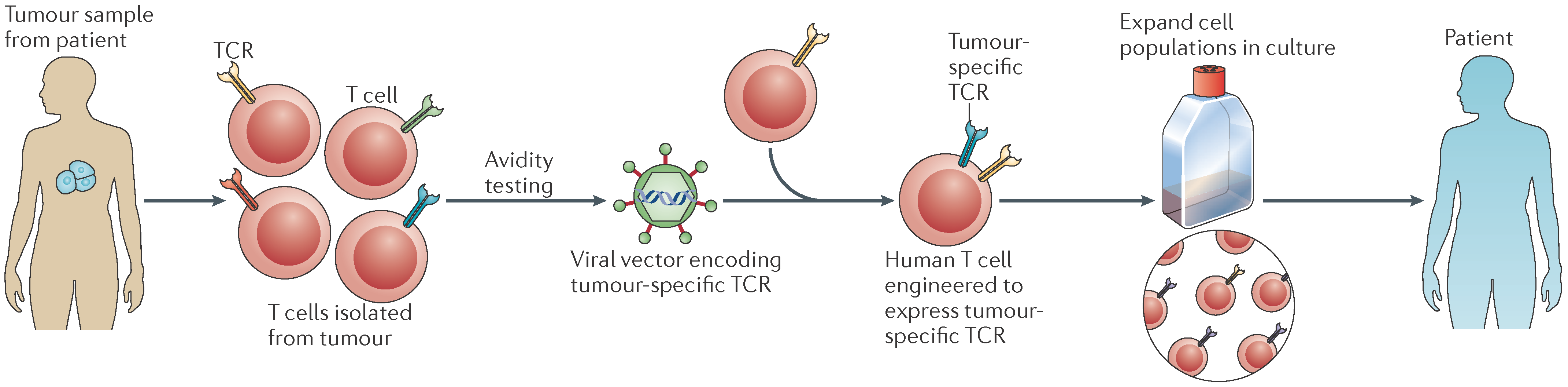
4.2. Adoptive Cell Therapy
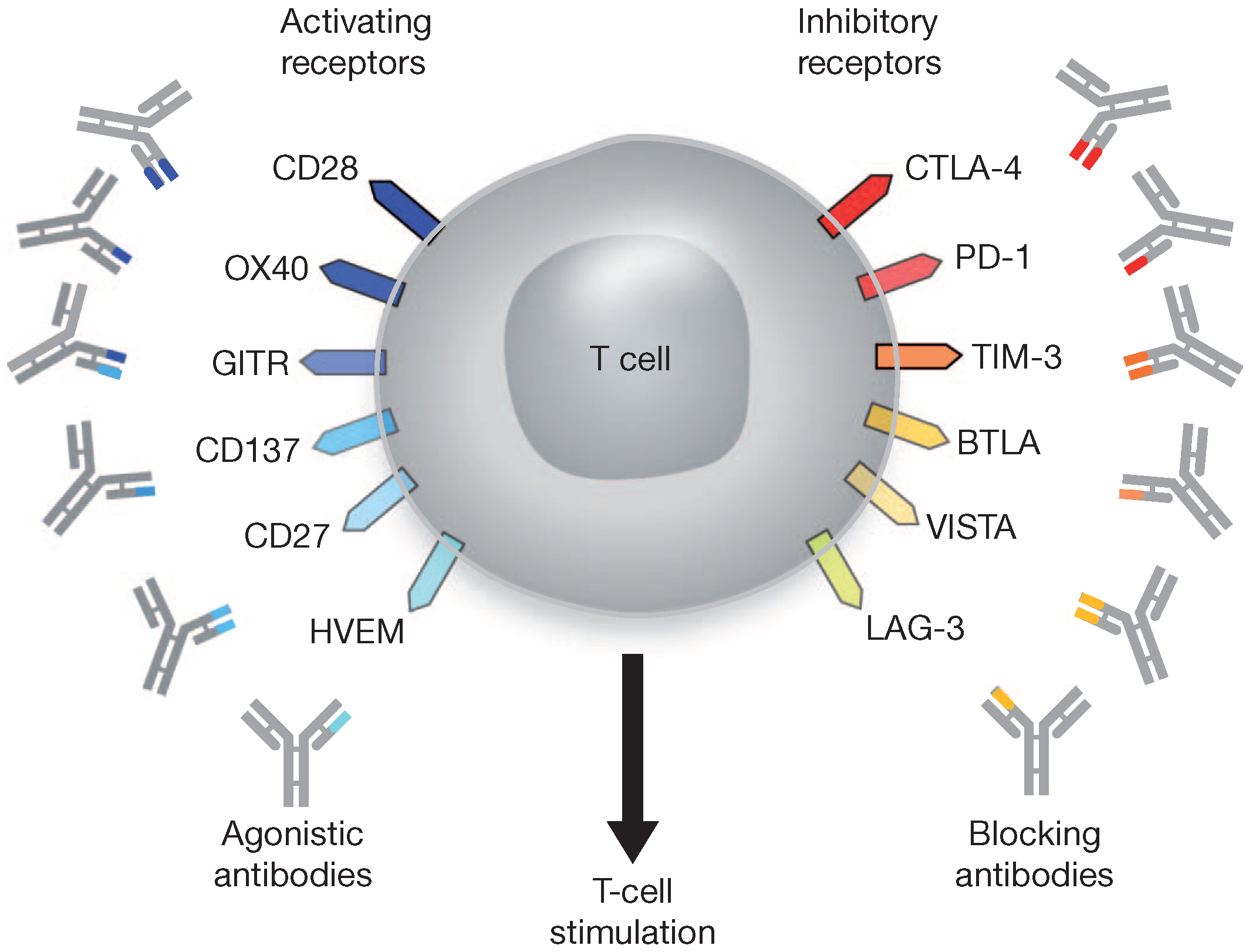
4.3. Immune Checkpoint Blockade
4.3.1. Programmed Death-1
4.3.2. CD160
4.3.3. Natural Killer Cell Receptor 2B4
4.3.4. Lymphocyte Activation Gene-3
4.3.5. T-Cell Immunoglobulin and Mucin-Domain Containing-3
4.3.6. Glycoprotein-2
4.4. Antiplatelet Therapy
5. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AFP | α-fetoprotein |
| APC | Antigen-presenting cell |
| BDCA-2 | Blood dendritic cell antigen-2 |
| CAR | Chimeric antigen receptor |
| CCA | Cholangiocarcinoma |
| CIK | Cytokine-induced killer cell |
| CTL | Cytotoxic T-lymphocyte |
| DC | Dendritic cell |
| GPC3 | Glypican-3 |
| GP-2 | Glycoprotein-2 |
| HBV | Hepatitis B virus |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HSC | Hepatic stellate cell |
| IFN-γ | Interferon-γ |
| IL-10 | Interleukin-10 |
| KC | Kupffer cell |
| LAG-3 | Lymphocyte activation gene-3 |
| LPS | Lipopolysaccharide |
| LSEC | Liver sinusoidal endothelial cell |
| MAGE-A | Melanoma antigen gene-A |
| MHC | Major histocompatibility complex |
| NAFLD | Non-alcoholic fatty liver disease |
| NK | Natural killer |
| NY-ESO-1 | New York esophageal squamous cell carcinoma 1 |
| NKT | NK T cell |
| PD-1 | Programmed death 1 |
| PD-L1 | Programmed death ligand 1 |
| PGE2 | Prostaglandin E2 |
| PSC | Primary sclerosing cholangitis |
| RFA | Radiofrequency ablation |
| SSX-2 | Synovial sarcoma X breakpoint 2 |
| TAA | Tumor-associated antigen |
| TAM | Tumor-associated macrophages |
| TAP-1 | Tapasin-1 |
| TCR | T-cell receptor |
| TERT | Telomerase reverse transcriptase |
| TGF-β | Transforming growth factor-β |
| TIL | Tumor-infiltrating lymphocyte |
| Tim-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TLR | Toll-like receptor |
| Treg | Regulatory T-cell |
References
- Nordenstedt, H.; White, D.L.; El-Serag, H.B. The changing pattern of epidemiology in hepatocellular carcinoma. Dig. Liver Dis. 2010, 42, S206–S214. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Steer, C.J.; Cressman, E.N. Molecular mechanisms of hepatocellular carcinoma. Hepatology 2008, 48, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, K.N.; Gores, G.J. Cholangiocarcinoma. Gastroenterology 2005, 128, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Bergquist, A.; Von Seth, E. Epidemiology of cholangiocarcinoma. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.R.; Oh, J.K.; Masuyer, E.; Curado, M.P.; Bouvard, V.; Fang, Y.Y.; Wiangnon, S.; Sripa, B.; Hong, S.T. Epidemiology of cholangiocarcinoma: An update focusing on risk factors. Cancer Sci. 2010, 101, 579–585. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Plummer, M.; Franceschi, S. Cholangiocarcinoma: Descriptive epidemiology and risk factors. Gastroenterol. Clin. Biol. 2010, 34, 173–180. [Google Scholar] [CrossRef]
- Shaib, Y.; El-Serag, H.B. The epidemiology of cholangiocarcinoma. Semin. Liver Dis. 2004, 24, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Eckmann, K.R.; Patel, D.K.; Landgraf, A.; Slade, J.H.; Lin, E.; Kaur, H.; Loyer, E.; Weatherly, J.M.; Javie, M. Chemotherapy outcomes for the treatment of unresectable intrahepatic and hilar cholangiocarcinoma: A retrospective analysis. Gastrointest. Cancer Res. 2011, 4, 155–160. [Google Scholar] [PubMed]
- Mancuso, A.; Perricone, G. Hepatocellular carcinoma and liver transplantation: State of the art. J. Clin. Transl. Hepatol. 2014, 2, 175–181. [Google Scholar]
- Sapisochín, G.; Fernández De Sevilla, E.; Echeverri, J.; Charco, R. Liver transplantation for cholangiocarcinoma: Current status and new insights. World J. Hepatol. 2015, 7, 2396–2403. [Google Scholar] [CrossRef]
- Crispe, I.N. The liver as a lymphoid organ. Ann. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Severi, T.; Van Malenstein, H.; Verslype, C.; Van Pelt, J.F. Tumor initiation and progression in hepatocellular carcinoma: Risk factors, classification, and therapeutic targets. Acta Pharmacol. Sin. 2010, 31, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wang, Z.; Lee, Y.; Chiang, Y.; Bonham, C.; Fung, J.; Lu, L. Hepatocyte-induced apoptosis of activated T cells, a mechanism of liver transplant tolerance, is related to the expression of ICAM-1 and hepatic lectin. Transp. Proc. 2001, 33, 226. [Google Scholar] [CrossRef]
- Bertolino, P.; Trescol-Biemont, M.C.; Rabourdin-Combe, C. Hepatocytes induce functional activation of naive CD8+ T lymphocytes but fail to promote survival. Eur. J. Immunol. 1998, 28, 221–236. [Google Scholar] [CrossRef]
- Bertolino, P.; McCaughan, G.W.; Bowen, D.G. Role of primary intrahepatic T-cell activation in the “liver tolerance effect”. Immunol. Cell Biol. 2002, 80, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Wuensch, S.A.; Spahn, J.; Crispe, I.N. Direct, help-independent priming of CD8+ T cells by adeno-associated virus-transduced hepatocytes. Hepatology 2010, 52, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di Lucia, P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the liver by intravascular effector CD8+ T cells. Cell 2015, 161, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.; Le Couteur, D.G.; Fraser, R.; Bowen, D.G.; McCaughan, G.W.; Bertolino, P. T lymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology 2006, 44, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Tabaczewski, P.; Truscott, S.M.; Van Kaer, L.; Stroynowski, I. Hepatocytes express abundant surface class I MHC and efficiently use transporter associated with antigen processing, tapasin, and low molecular weight polypeptide proteasome subunit components of antigen processing and presentation pathway. J. Immunol. 2005, 175, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, R.; Kanda, T.; Wu, S.; Nakamoto, S.; Haga, Y.; Jiang, X.; Nakamura, M.; Shirasawa, H.; Yokosuka, O. Association between hepatitis B virus and MHC class I polypeptide-related chain A in human hepatocytes derived from human-mouse chimeric mouse liver. Biochem. Biophys. Res. Commun. 2015, 464, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Herkel, J.; Jagemann, B.; Wiegard, C.; Lazaro, J.F.; Lueth, S.; Kanzler, S.; Blessing, M.; Schmitt, E.; Lohse, A.W. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocytes. Hepatology 2003, 37, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Liver antigen-presenting cells. J. Hepatol. 2011, 54, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.H.; Tucker, R.M.; Wehrmann, F.; Mack, D.G.; Ueno, Y.; Mack, C.L. Cholangiocytes as immune modulators in rotavirus-induced murine biliary atresia. Liver Int. 2009, 29, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Heydtmann, M.; Lalor, P.F.; Eksteen, J.A.; Hübscher, S.G.; Briskin, M.; Adams, D.H. CXC chemokine ligand 16 promotes integrin-mediated adhesion of liver-infiltrating lymphocytes to cholangiocytes and hepatocytes within the inflamed human liver. J. Immunol. 2005, 174, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Callery, M.P.; Mangino, M.J.; Flye, M.W. Arginine-specific suppression of mixed lymphocyte culture reactivity by Kupffer cells—A basis of portal venous tolerance. Transplantation 1991, 51, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.; Schlaak, J.; Uhrig, A.; Kempf, P.; Meyer zum Büschenfelde, K.H.; Gerken, G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J. Hepatol. 1995, 22, 226–229. [Google Scholar] [CrossRef]
- You, Q.; Cheng, L.; Kedl, R.M.; Ju, C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 2008, 48, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Gerken, G. Local control of the immune response in the liver. Immunol. Rev. 2000, 174, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Maemura, K.; Zheng, Q.; Wada, T.; Ozaki, M.; Takao, S.; Aikou, T.; Bulkley, G.B.; Klein, A.S.; Sun, Z. Reactive oxygen species are essential mediators in antigen presentation by Kupffer cells. Immunol. Cell Biol. 2005, 83, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-Y.; Moriarty, T.J.; Wong, C.H.Y.; Zhou, H.; Strieter, R.M.; Van Rooijen, N.; Chaconas, G.; Kubes, P. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat. Immunol. 2010, 11, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Goddard, S.; Youster, J.; Morgan, E.; Adams, D.H. Interleukin-10 secretion differentiates dendritic cells from human liver and skin. Am. J. Pathol. 2004, 164, 511–519. [Google Scholar] [CrossRef]
- Tokita, D.; Sumpter, T.L.; Raimondi, G.; Zahorchak, A.F.; Wang, Z.; Nakao, A.; Mazariegos, G.V.; Abe, M.; Thomson, A.W. Poor allostimulatory function of liver plasmacytoid DC is associated with pro-apoptotic activity, dependent on regulatory T cells. J. Hepatol. 2008, 49, 1008–1018. [Google Scholar]
- Kingham, T.P.; Chaudhry, U.I.; Plitas, G.; Katz, S.C.; Raab, J.; DeMatteo, R.P. Murine liver plasmacytoid dendritic cells become potent immunostimulatory cells after Flt-3 ligand expansion. Hepatology 2007, 45, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.K.; Bedrosian, A.S.; Malhotra, A.; Henning, J.R.; Ibrahim, J.; Vera, V.; Cieza-Rubio, N.E.; Hassan, B.U.; Pachter, H.L.; Cohen, S.; et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J. Immunol. 2010, 185, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Elvevold, K.; Smedsrød, B.; Martinez, I. The liver sinusoidal endothelial cell: A cell type of controversial and confusing identity. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G391–G400. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Germann, T.; Treichel, U.; Uhrig, A.; Schmitt, E.; Hegenbarth, S.; Lohse, A.W.; Gerken, G. Endotoxin down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells. J. Immunol. 1999, 162, 1401–1407. [Google Scholar]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Winau, F.; Hegasy, G.; Weiskirchen, R.; Weber, S.; Cassan, C.; Sieling, P.A.; Modlin, R.L.; Liblau, R.S.; Gressner, A.M.; Kaufmann, S.H. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 2007, 26, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Bomble, M.; Tacke, F.; Rink, L.; Kovalenko, E.; Weiskirchen, R. Analysis of antigen-presenting functionality of cultured rat hepatic stellate cells and transdifferentiated myofibroblasts. Biochem. Biophys. Res. Commun. 2010, 396, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Eksteen, B.; Mora, J.R.; Haughton, E.L.; Henderson, N.C.; Lee-Turner, L.; Villablanca, E.J.; Curbishley, S.M.; Aspinall, A.I.; Von Andrian, U.H.; Adams, D.H. Gut homing receptors on CD8 T cells are retinoic acid dependent and not maintained by liver dendritic or stellate cells. Gastroenterology 2009, 137, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Criscitiello, C.; Esposito, A.; Trapani, D.; Curigliano, G. Prognostic and predictive value of tumor infiltrating lymphocytes in early breast cancer. Cancer Treat. Rev. 2016, 50, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Shirabe, K.; Motomura, T.; Muto, J.; Toshima, T.; Matono, R.; Mano, Y.; Takeishi, K.; Ijichi, H.; Harada, N.; Uchiyama, H.; et al. Tumor-infiltrating lymphocytes and hepatocellular carcinoma: Pathology and clinical management. Int. J. Clin. Oncol. 2010, 15, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Knief, J.; Reddemann, K.; Petrova, E.; Herhahn, T.; Wellner, U.; Thorns, C. High density of tumor-infiltrating B-lymphocytes and plasma cells signifies prolonged overall survival in adenocarcinoma of the esophagogastric junction. Anticancer Res. 2016, 36, 5339–5345. [Google Scholar] [CrossRef] [PubMed]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Breous, E.; Thimme, R. Potential of immunotherapy for hepatocellular carcinoma. J. Immunol. 2011, 54, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Allavena, P.; Garlanda, C.; Locati, M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum. Immunol. 2009, 70, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Germano, G.; Marchesi, F.; Locatelli, M.; Biswas, S.K. Cancer-promoting tumor-associated macrophages: New vistas and open questions. Eur. J. Immunol. 2011, 41, 2522–2525. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stangé, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C (high) monocytes. Cancer Res. 2010, 71, 5728–5739. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Candido, S.; Cervello, M.; Nicolosi, D.; Raiti, F.; Travali, S.; Spandidos, D.A.; Libra, M. The tumor microenvironment in hepatocellular carcinoma (review). Int. J. Oncol. 2012, 40, 1733–1747. [Google Scholar] [PubMed]
- Ramaiah, S.K.; Rittling, S. Pathophysiological role of osteopontin in hepatic inflammation, toxicity, and cancer. Toxicol. Sci. 2008, 103, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Pierantenelli, I.; Banales, J.M.; Benedetti, A.; Marzioni, M. The significance of genetics for cholangiocarcinoma development. Ann. Transl. Med. 2013, 1, 3. [Google Scholar]
- Szabo, G.; Dolganiuc, A.; Mandrekar, P. Pattern recognition receptors: A contemporary view on liver diseases. Hepatology 2006, 44, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gallo, D.J.; Green, A.M.; Williams, D.L.; Gong, X.; Shapiro, R.A.; Gambotto, A.A.; Humphris, E.L.; Vodovotz, Y.; Billiar, T.R. Role of Toll-like receptors in changes in gene expression and NF-κB activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect. Immun. 2002, 70, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Ito, A.; Takii, T.; Hayashi, H.; Onozaki, K. Endotoxin and cytokine regulation of Toll-like receptor (TLR) 2 and TLR4 gene expression in murine liver and hepatocytes. J. Interferon Cytokine Res. 2000, 201, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Meng, Z.; Jiang, M.; Zhang, E.; Trippler, M.; Broering, R.; Bucchi, A.; Krux, F.; Dittmer, U.; Yang, D.; et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 2010, 129, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Martin-Armas, M.; Simon-Santamaria, J.; Pettersen, I.; Moens, U.; Smedsrød, B.; Sveinbjørnsson, B. Toll-like receptor 9 (TLR9) is present in murine liver sinusoidal endothelial cells (LSECs) and mediates the effect of CpG-oligonucleotides. J. Hepatol. 2006, 44, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, G.L.; Klein, R.D.; Aminlari, A.; Zhang, H.Y.; Steinstraesser, L.; Alarcon, W.H.; Remick, D.G.; Wang, S.C. Kupffer cell activation by lipopolysaccharide in rats: Role for lipopolysaccharide binding protein and Toll-like receptor 4. Hepatology 2000, 31, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Thobe, B.M.; Frink, M.; Hildebrand, F.; Schwacha, M.G.; Hubbard, W.J.; Choudhry, M.A.; Chaudry, I.H. The role of MAPK in Kupffer cell Toll-like receptor (TLR) 2-, TLR4-, and TLR9-mediated signaling following trauma-hemorrhage. J. Cell Physiol. 2007, 210, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β-signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Hashmi, A.; Gomes, D.A.; Town, T.; Badou, A.; Flavell, R.A.; Mehal, W.Z. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 2007, 46, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R.N. Role of innate immunity in the development of hepatocellular carcinoma. World J. Gastroenterol. 2013, 19, 7500–7514. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, H.; Suzuki, H.; Shimomura, M.; Kojima, M.; Gotohda, N.; Takahashi, S.; Nakagohri, T.; Konishi, M.; Kobayashi, N.; Kinoshita, T.; et al. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2009, 100, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Yoshikawa, T.; Nobuoka, D.; Shirakawa, H.; Kuronuma, T.; Motomura, Y.; Mizuno, S.; Ishii, H.; Nakachi, K.; Konishi, M.; et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: Immunologic evidence and potential for improving overall survival. Clin. Cancer Res. 2012, 18, 3686–3696. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Yoshikawa, T.; Ofuji, K.; Yoshimura, M.; Tsuchiya, N.; Takahashi, M.; Nobuoka, D.; Gotohda, N.; Takahashi, S.; Kato, Y.; et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology 2016, 5, e1129483. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Forner, A.; Korangy, F.; N′Kontchou, G.; Barget, N.; Ayuso, C.; Ormandy, L.A.; Manns, M.P.; Beaugrand, M.; Bruix, J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 2010, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Lam, S.S.; Liu, D.; Kim, D.Y.; Ma, L.; Alleruzzo, L.; Chen, W.; Hode, T.; Henry, C.J.; Kaifi, J.; et al. Development of InCVAX, in situ cancer vaccine, and its immune response in mice with hepatocellular cancer. J. Clin. Cell Immunol. 2016, 7, 438. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Li, X.; Naylor, M.F.; Hode, T.; Nordquist, R.E.; Alleruzzo, L.; Raker, J.; Lam, S.S.; Du, N.; Shi, L.; et al. InCVAX—A novel strategy for treatment of late-stage, metastatic cancers through photoimmunotherapy induced tumor-specific immunity. Cancer Lett. 2015, 359, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-P.; Wang, Q.-X.; Lin, H.-P.; Xu, B.; Zhao, Q.; Chen, K. Recombinant heat shock protein 70 functional peptide and alpha-fetoprotein epitope peptide vaccine elicits specific anti-tumor immunity. Oncotarget 2016, 7, 71274–71284. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Kotera, Y.; Aruga, A.; Takeshita, N.; Takasaki, K.; Yamamoto, M. Clinical utilization of postoperative dendritic cell vaccine plus activated T-cell transfer in patients with intrahepatic cholangiocarcinoma. J. Hepatobiliary Pancreat Sci. 2012, 19, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, N.; Yamamoto, M.; Aruga, A.; Takasaki, K.; Nakano, M. Correlation between expression of MUC1 core protein and outcome after surgery in mass-forming intrahepatic cholangiocarcinoma. Cancer 2002, 94, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Mall, A.S.; Tyler, M.G.; Ho, S.B.; Krige, J.E.; Kahn, D.; Spearman, W.; Myer, L.; Govender, D. The expression of MUC mucin in cholangiocarcinoma. Pathol. Res. Pract. 2010, 206, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Boonla, C.; Sripa, B.; Thuwajit, P.; Cha-On, U.; Puapairoj, A.; Miwa, M.; Wongkham, S. MUC1 and MUC5AC mucin expression in liver fluke-associated intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2005, 11, 4939–4946. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Sakabe, T.; Abe, H.; Tanii, M.; Takahashi, H.; Chiba, A.; Yanagida, E.; Shibamoto, Y.; Ogasawara, M.; Tsujitani, S.; et al. Dendritic cell-based immunotherapy targeting synthesized peptides for advanced biliary tract cancer. J. Gastrointest. Surg. 2013, 17, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, N.; Okamoto, S.; Amaishi, Y.; Sato, E.; Seo, N.; Mineno, J.; Takesako, K.; Kato, T.; Shiku, H. Efficient tumor regression by adoptively transferred CEA-specific CAR-T cells associated with symptoms of mild cytokine release syndrome. Oncoimmunology 2016, 5, e1211218. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Li, K.; Tu, H.; Pan, X.; Jiang, H.; Shi, B.; Kong, J.; Wang, H.; Yang, S.; Gu, J.; et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 6418–6428. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Sleijfer, S.; Van Steenbergen, S.; Van Elzakker, P.; Van Krimpen, B.; Groot, C.; Vulto, A.; Den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Riley, J.; Dudley, M.E.; Rosenberg, S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but dose not mediate tumor regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Herlyn, D. Combination of active specific immunotherapy or adoptive antibody or lymphocyte immunotherapy with chemotherapy in the treatment of cancer. Cancer Immunol. Immunother. 2009, 58, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, M.; Pievani, A.; Borleri, G.; Vago, L.; Fleischhauer, K.; Golay, J.; Introna, M. Cytokine-induced killer cells are terminally differentiated activated CD8 cytotoxic T-EMRA lymphocytes. Exp. Hematol. 2009, 37, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.; Qiang, L.; Jian, W.; Ti, Z.; Da-Lu, K. A randomized, controlled trial of postoperative adjuvant cytokine-induced killer cells immunotherapy after radical resection of hepatocellular carcinoma. Dig. Liver Dis. 2009, 41, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Sekine, T.; Makuuchi, M.; Yamasaki, S.; Kosuge, T.; Yamamoto, J.; Shimada, K.; Sakamoto, M.; Hirohashi, S.; Ohashi, Y.; et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: A randomised trial. Lancet 2000, 356, 802–807. [Google Scholar] [CrossRef]
- Ma, Y.; Xu, Y.C.; Tang, L.; Zhang, Z.; Wang, J.; Wang, H.X. Cytokine-induced killer (CIK) cell therapy for patients with hepatocellular carcinoma: Efficacy and safety. Exp. Hematol. Oncol. 2012, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J.; Gwak, G.Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015, 148, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.S.; Zhou, J.; Zhou, Q.M.; Zhao, M.; Wang, Q.J.; Huang, L.X.; Li, Y.Q.; Chen, S.P.; Wu, P.H.; Xia, J.C. Minimally invasive treatment combined with cytokine-induced killer cells therapy lower the short-term recurrence rates of hepatocellular carcinomas. J. Immunother. 2008, 31, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhao, H.; Liu, L.; Cao, S.; Ren, B.; Zhang, N.; An, X.; Yu, J.; Li, H.; Ren, X. A randomized phase II study of autologous cytokine-induced killer cells in treatment of hepatocellular carcinoma. J. Clin. Immunol. 2014, 34, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Wongkajornsilp, A.; Somchitprasert, T.; Butraporn, R.; Wamanuttajinda, V.; Kasetsinsombat, K.; Huabprasert, S.; Maneechotesuwan, K.; Hongeng, S. Human cytokine-induced killer cells specifically infiltrated and retarded the growth of the inoculated human cholangiocarcinoma cells in SCID mice. Cancer Investig. 2009, 27, 140–148. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, Y.; Shi, W.J.; Wang, S.J.; Sha, H.F.; Feng, J.X.; Wang, J. Immunomodulation of inducible co-stimulator (ICOS) in human cytokine-induced killer cells against cholangiocarcinoma through ICOS/ICOS ligand interaction. J. Dig. Dis. 2011, 12, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, T.; Umebayashi, M.; Kiyota, A.; Koya, N.; Tanaka, H.; Onishi, H.; Katano, M. Combining cetuximab with killer lymphocytes synergistically inhibits human cholangiocarcinoma cells in vitro. Anticancer Res. 2012, 32, 2249–2256. [Google Scholar] [PubMed]
- Löffler, M.W.; Chandran, P.A.; Laske, K.; Schroeder, C.; Bonzheim, I.; Walzer, M.; Hilke, F.J.; Trautwein, N.; Kowalewski, D.J.; Schuster, H.; et al. Personalized peptide vaccine-induced immune response associated with long-term survival of a metastatic cholangiocarcinoma patient. J. Hepatol. 2016, 65, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Freeman, G.J.; McDermott, D.F. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin. Ther. 2015, 37, 764–782. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lee, H.T.; Shin, W.; Chae, J.; Choi, J.; Kim, S.H.; Lim, H.; Won Heo, T.; Park, K.Y.; Lee, Y.J.; et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun. 2016, 7, 13354. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kefford, R.; Carlino, M. PD-1 and PD-L1 inhibitors in melanoma treatment: Past success, present application and future challenges. Immunotherapy 2016, 8, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Specenier, P. Ipilimumab in melanoma. Expert Rev. Anticancer Ther. 2016, 16, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Gomez-Martin, C.; De la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Hiroishi, K.; Eguchi, J.; Baba, T.; Shimazaki, T.; Ishii, S.; Hiraide, A.; Sakaki, M.; Doi, H.; Uozumi, S.; Omori, R.; et al. Strong CD8+ T-cell responses against tumor-associated antigens prolong the recurrence-free interval after tumor treatment in patients with hepatocellular carcinoma. J. Gastroenterol. 2010, 45, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.; Lalezari, J.; Lawitz, E.; DiMicco, M.; Ghalib, R.; Reddy, K.R.; Chang, K.M.; Sulkowski, M.; Marro, S.O.; Anderson, J.; et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS ONE 2013, 8, e63818. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Nam, A.R.; Bang, J.H.; Park, J.E.; Kim, T.Y.; Lee, K.H.; Han, S.W.; Im, S.A.; Kim, T.Y.; Bang, Y.J.; et al. Soluble programmed death-ligand 1 (sPDL1) and neutrophil-to-lymphocyte ratio (NLR) predicts survival in advanced biliary tract cancer patients treated with palliative chemotherapy. Oncotarget 2016, 7, 76604–76612. [Google Scholar] [CrossRef] [PubMed]
- Sabbatino, F.; Villani, V.; Yearley, J.H.; Deshpande, V.; Cai, L.; Konstantinidis, I.T.; Moon, C.; Nota, S.; Wang, Y.; Al-Sukaini, A.; et al. PD-L1 and HLA class 1 antigen expression and clinical course of the disease in intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2016, 22, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Schlaphoff, V.; Lunemann, S.; Suneetha, P.V.; Jaroszewicz, J.; Grabowski, J.; Dietz, J.; Helfritz, F.; Bektas, H.; Sarrazin, C.; Manns, M.P.; et al. Dual function of the NK cell receptor 2B4 (CD244) in the regulation of HCV-specific CD8+ T cells. PLoS Pathog. 2011, 7, e1002045. [Google Scholar] [CrossRef] [PubMed]
- Kroy, D.C.; Ciuffreda, D.; Cooperrider, J.H.; Tomlinson, M.; Hauck, G.D.; Aneja, J.; Berger, C.; Wolski, D.; Carrington, M.; Wherry, E.J.; et al. Liver environment and HCV replication affect human T-cell phenotype and expression of inhibitory receptors. Gastroenterology 2014, 146, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.H.; Boles, K.; Van der Merwe, P.A.; Kumar, V.; Mathew, P.A.; Barclay, A.N. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J. Exp. Med. 1998, 188, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Liu, Y.; Guo, Y.; Chen, Y.; Liu, X.; Liu, M. Lymphocyte activation gene 3 negatively regulates the function of intrahepatic hepatitis C virus-specific CD8+ T cells. J. Gastroenterol. Hepatol. 2015, 30, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Zhang, Y.; Jin, G.X.; Yao, L.; Wu, D.Q. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8+ T cell in HCC patients. Immunol. Lett. 2013, 150, 116–122. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- McMahan, R.H.; Golden-Mason, L.; Nishimura, M.I.; McMahon, B.J.; Kemper, M.; Allen, T.M.; Gretch, D.R.; Rosen, H.R. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J. Clin. Investig. 2010, 120, 4546–4557. [Google Scholar] [CrossRef] [PubMed]
- Jendrek, S.T.; Gotthardt, D.; Nitzsche, T.; Widmann, L.; Korf, T.; Michaels, M.A.; Weiss, K.H.; Liaskou, E.; Vesterhus, M.; Karlsen, T.H.; et al. Anti-GP2 IgA autoantibodies are associated with poor survival and cholangiocarcinoma in primary sclerosing cholangitis. Gut 2016, 66, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Erdal, E.; Atabey, N.; Carr, B.I. Platelets, microenvironment and hepatocellular carcinoma. Biochem. Anal. Biochem. 2016, 5, 281. [Google Scholar] [CrossRef]
- Kurokawa, T.; Zheng, Y.W.; Ohkohchi, N. Novel functions of platelets in the liver. J. Gastroenterol. Hepatol. 2016, 31, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.J.; Luo, J.C.; Li, C.P.; Chu, C.W.; Wu, J.C.; Lai, C.R.; Chiang, J.H.; Chau, G.Y.; Lui, W.Y.; Lee, C.C.; et al. Thrombocytosis: A paraneoplastic syndrome in patients with hepatocellular carcinoma. World J. Gastroenterol. 2004, 10, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lin, Y.J.; Lin, C.C.; Yen, C.L.; Shen, C.H.; Chang, C.J.; Hsieh, S.Y. Pretreatment platelet count early predicts extrahepatic metastasis of human hepatoma. Liver Int. 2015, 35, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Araki, K.; Hirai, K.; Kubo, N.; Igarashi, T.; Tsukagoshi, M.; Ishii, N.; Hoshino, K.; Kuwano, H.; Shirabe, K. A novel clinical factor, D-dimer platelet multiplication, may predict postoperative recurrence and prognosis for patients with cholangiocarcinoma. Ann. Surg. Oncol. 2016, 23, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.N.; Wang, J.H.; Liu, S.L.; Hung, C.H.; Chen, C.H.; Tung, H.D.; Chen, T.M.; Huang, W.S.; Lee, C.M.; Chen, C.C.; et al. Thrombocytopenia as a surrogate for cirrhosis and a marker for the identification of patients at high-risk for hepatocellular carcinoma. Cancer 2006, 107, 2212–2222. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Qu, K.; Zhang, J.Y.; Song, S.D.; Liu, S.S.; Tai, M.H.; Liu, H.C.; Liu, C. The prognostic value of platelet count in patients with hepatocellular carcinoma: A systematic review and meta-analysis. Medicine 2015, 94, e1431. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Marchese, P.; Castro, M.G.; Lowenstein, P.R.; Chisari, F.V.; Ruggeri, Z.M.; Guidotti, L.G. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat. Med. 2005, 11, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Sitia, G.; Aiolfi, R.; Di Lucia, P.; Mainetti, M.; Fiocchi, A.; Mingozzi, F.; Esposito, A.; Ruggeri, Z.M.; Chisari, F.V.; Iannacone, M.; et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc. Natl. Acad. Sci. USA 2012, 109, E2165–E2172. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Yeh, C.M.; Hu, Y.W.; Chen, C.C.; Liu, C.J.; Su, C.W.; Huo, T.I.; Huang, Y.H.; Chao, Y.; Chen, T.J.; et al. Antiplatelet therapy is associated with a better prognosis for patients with hepatitis B virus-related hepatocellular carcinoma after liver resection. Ann. Surg. Oncol. 2016, 23, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Schurich, A. Platelets harness the immune response to drive liver cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 12840–12841. [Google Scholar] [CrossRef] [PubMed]
- McMorran, B.J.; Marshall, V.M.; De Graaf, C.; Drysdale, K.E.; Shabbar, M.; Smyth, G.K.; Corbin, J.E.; Alexander, W.S.; Foote, S.J. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science 2009, 323, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Sitia, G.; Iannacone, M.; Guidotti, L.G. Anti-platelet therapy in the prevention of hepatitis b virus-associated hepatocellular carcinoma. J. Hepatol. 2013, 59, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Thein, H.H.; Isaranuwatchai, W.; Campitelli, M.A.; Feld, J.J.; Yoshida, E.; Sherman, M.; Hoch, J.S.; Peacock, S.; Krahn, M.D.; Earle, C.C. Health care costs associated with hepatocellular carcinoma: A population-based study. Hepatology 2013, 58, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Tapper, E.B.; Catana, A.M.; Sethi, N.; Mansuri, D.; Sethi, S.; Vong, A.; Afdhal, N.H. Direct costs of care for hepatocellular carcinoma in patients with hepatitis C cirrhosis. Cancer 2016, 122, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, N.; Sawada, Y.; Endo, I.; Uemura, Y.; Nakatsura, T. Potentiality of immunotherapy against hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 10314–10326. [Google Scholar] [CrossRef] [PubMed]
- Tampellini, M.; La Salvia, A.; Scagliotti, G.V. Novel investigational therapies for treating biliary tract carcinoma. Expert Opin. Investig. Drugs 2016, 25, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Feng, M.; Kim, H.; Phung, Y.; Kleiner, D.E.; Gores, G.J.; Qian, M.; Wang, X.W.; Ho, M. Mesothelin as a potential therapeutic target in human cholangiocarcinoma. J. Cancer 2010, 1, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Hünig, T. The rise and fall of the CD28 superagonist TGN1412 and its return as TAB08: A personal account. FASEB J. 2016, 283, 3325–3334. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Immunotherapeutic Strategy | Mode of Application | Mechanism of Action |
|---|---|---|
| Vaccines | Antigenic peptides/proteins (GPC-3, AFP, NDGC, MUC1) DC-based vaccines, APCs from tumor lysates, InCVAX | Targeting TAAs to overcome immune tolerance by expressing antigenic proteins/peptides or co-stimulatory molecules in DCs or tumor cells |
| Adoptive cell therapy | CIK infusion, CTL transfer TCR-T cells | Transfer of tumor-specific T cells from a healthy individual into the bloodstream of the patient to be treated after propagating them ex vivo to enhance immune responses |
| CAR-T cells, TCR-T cells | Patient-derived T cells are modified to express artificial receptors, propagated in vitro and administered back into the same patient | |
| Immune checkpoint blockade | Antibody (Pembrolizumab Ipilimumab, Nivolumab Tremelimumab, AMP-224 Lambrolizumab, CT-011, and others) | Targeting specific cellular receptors and their ligands (PD-1, PD-L1, CTLA-1, CD160, 2B4, LAG-3, Tim-3, GP-2), and to enhance antigen presentation |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aravalli, R.N.; Steer, C.J. Immune-Mediated Therapies for Liver Cancer. Genes 2017, 8, 76. https://doi.org/10.3390/genes8020076
Aravalli RN, Steer CJ. Immune-Mediated Therapies for Liver Cancer. Genes. 2017; 8(2):76. https://doi.org/10.3390/genes8020076
Chicago/Turabian StyleAravalli, Rajagopal N., and Clifford J. Steer. 2017. "Immune-Mediated Therapies for Liver Cancer" Genes 8, no. 2: 76. https://doi.org/10.3390/genes8020076
APA StyleAravalli, R. N., & Steer, C. J. (2017). Immune-Mediated Therapies for Liver Cancer. Genes, 8(2), 76. https://doi.org/10.3390/genes8020076