Deep Transcriptome Sequencing of Two Green Algae, Chara vulgaris and Chlamydomonas reinhardtii, Provides No Evidence of Organellar RNA Editing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Culturing
2.2. Deep Sequencing and Genome Assembly
2.3. RNA Editing Detection
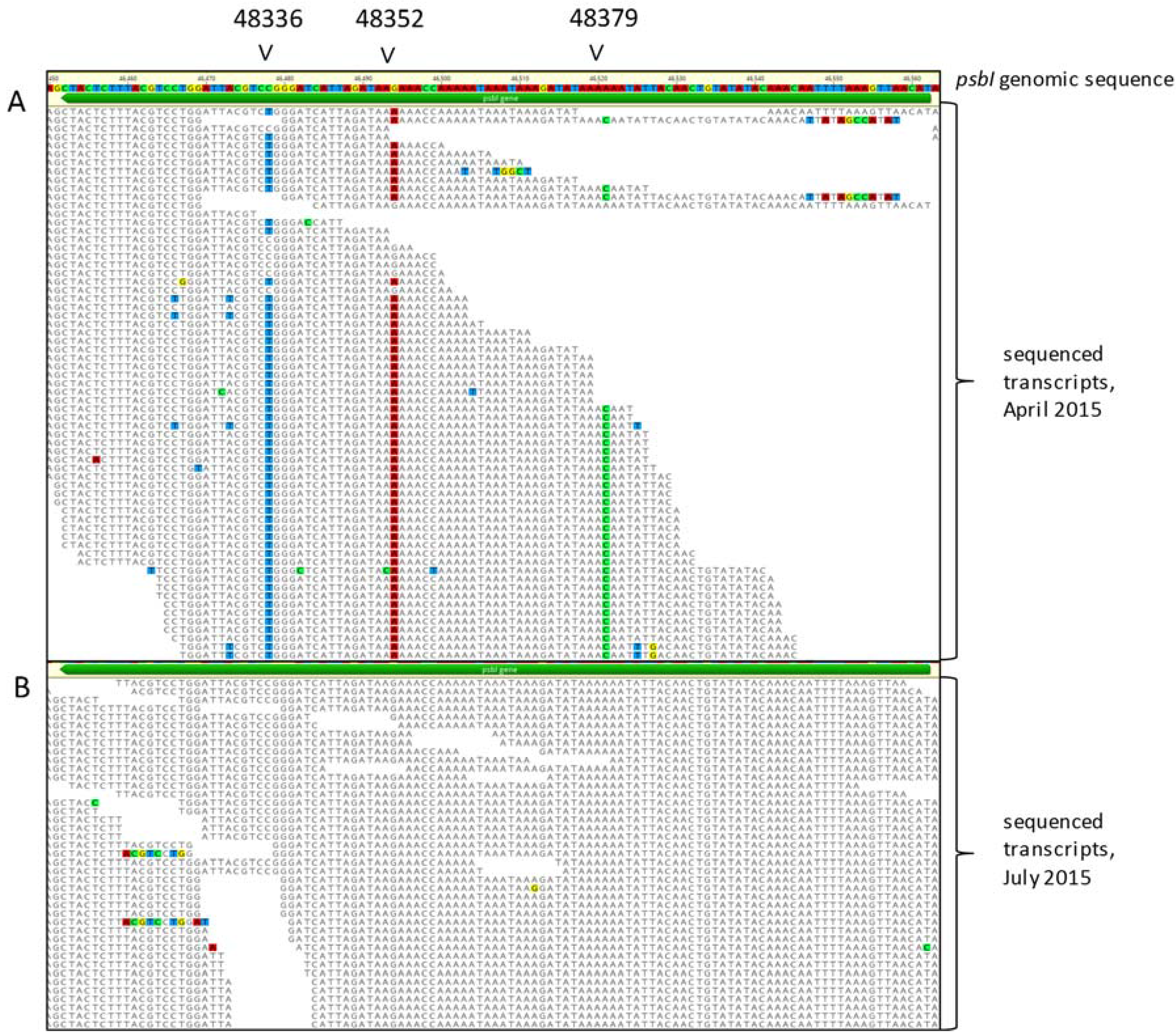
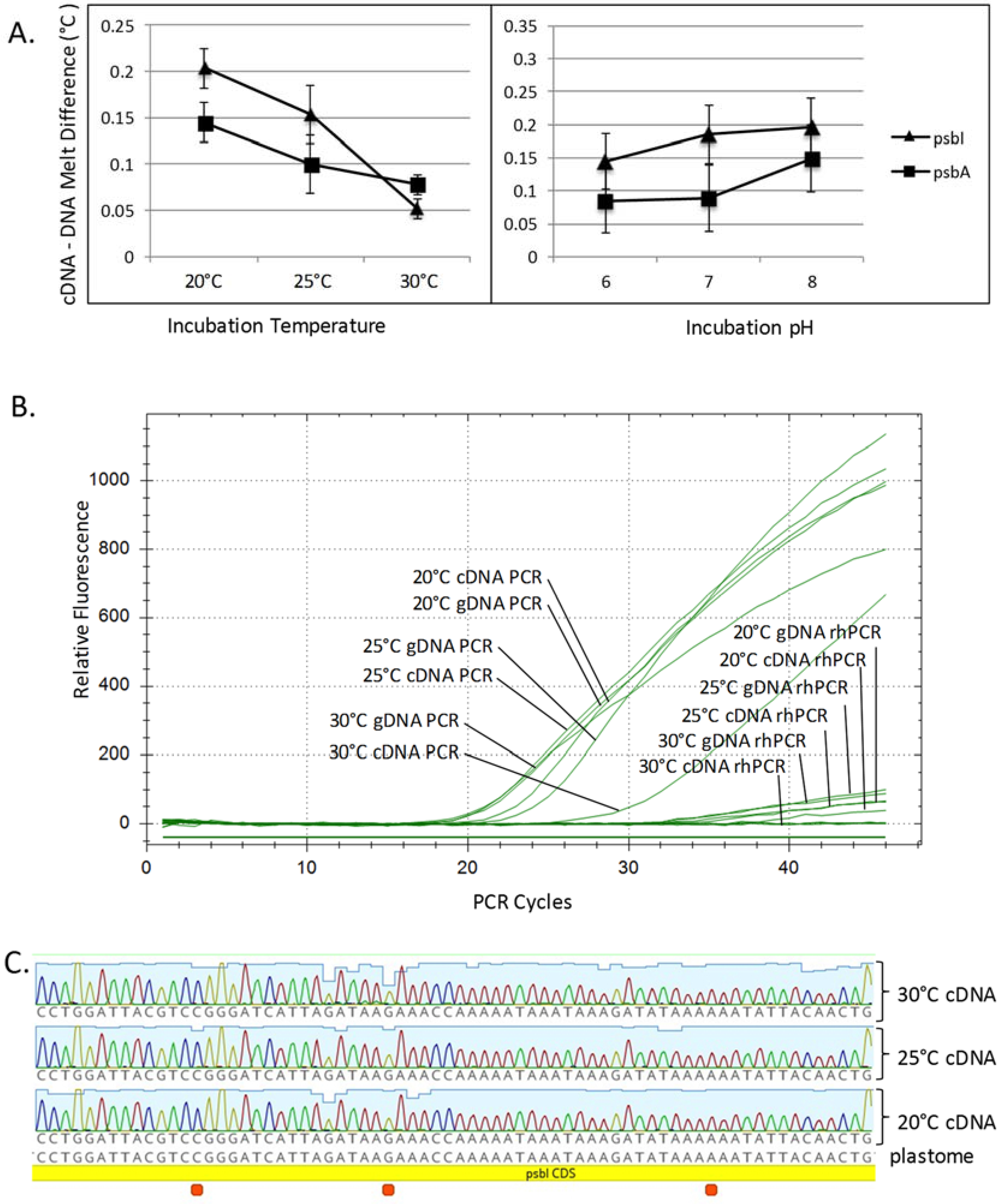
2.4. High Resolution Melt Analysis of Chara psbI
2.5. RNase H-Dependent PCR Analysis of Chara psbI
2.6. Sanger Sequencing
3. Results
3.1. C. vulgaris Organellar Genomes
3.2. C. vulgaris Transcriptome RNA Editing Screen
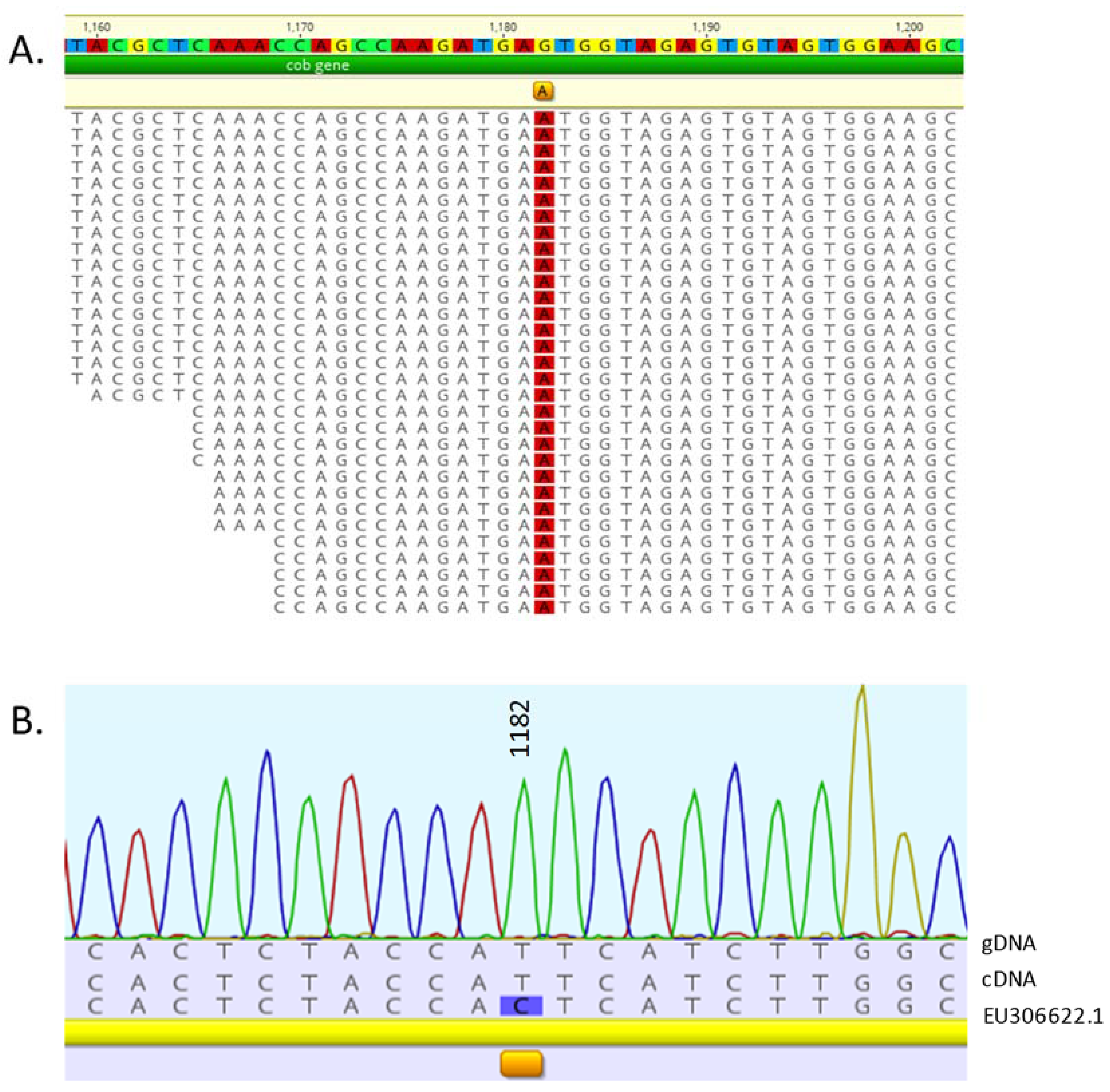
3.3. C. reinhardtii Transcriptome RNA Editing Screen
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Freyer, R.; Kiefer-Meyer, M.C.; Kossel, H. Occurrence of plastid RNA editing in all major lineages of land plants. Proc. Natl. Acad. Sci. USA 1997, 94, 6285–6290. [Google Scholar] [CrossRef] [PubMed]
- Covello, P.S.; Gray, M.W. RNA editing in plant mitochondria. Nature 1989, 341, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Lamattina, L.; Bonnard, G.; Weil, J.H.; Grienenberger, J.M. RNA editing in wheat mitochondria results in the conservation of protein sequences. Nature 1989, 341, 660–662. [Google Scholar] [CrossRef] [PubMed]
- Hiesel, R.; Wissinger, B.; Schuster, W.; Brennicke, A. RNA editing in plant mitochondria. Science 1989, 246, 1632–1634. [Google Scholar] [CrossRef] [PubMed]
- Bruhs, A.; Kempken, F. RNA Editing in Higher Plant Mitochondria. In Plant Mitochondria; Springer: New York, NY, USA, 2011; pp. 157–175. [Google Scholar]
- Kempken, F.; Hofken, G.; Pring, D.R. Analysis of silent RNA editing sites in atp6 transcripts of Sorghum bicolor. Curr. Genet. 1995, 27, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Grimes, B.T.; Sisay, A.K.; Carroll, H.D.; Cahoon, A.B. Deep sequencing of the tobacco mitochondrial transcriptome reveals expressed ORFs and numerous editing sites outside coding regions. BMC Genomics 2014, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Unseld, M.; Marienfeld, J.R.; Brandt, P.; Brennicke, A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 1997, 15, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Binder, S.; Marchfelder, A.; Brennicke, A.; Wissinger, B. RNA editing in trans-splicing intron sequences of nad2 mRNAs in Oenothera mitochondria. J. Biol. Chem. 1992, 267, 7615–7623. [Google Scholar] [PubMed]
- Börner, G.V.; Mörl, M.; Wissinger, B.; Brennicke, A.; Schmelzer, C. RNA editing of a group II intron in Oenothera as a prerequisite for splicing. Mol. Gen. Genet. 1995, 246, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Morton, B.R.; Maier, U.G. The evolution of chloroplast RNA editing. Mol. Biol. Evol. 2006, 23, 1912–1921. [Google Scholar] [CrossRef] [PubMed]
- Groth-Malonek, M.; Wahrmund, U.; Polsakiewicz, M.; Knoop, V. Evolution of a pseudogene: Exclusive survival of a functional mitochondrial nad7 gene supports Haplomitrium as the earliest liverwort lineage and proposes a secondary loss of RNA editing in Marchantiidae. Mol. Biol. Evol. 2007, 24, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Salone, V.; Rüdinger, M.; Polsakiewicz, M.; Hoffmann, B.; Groth-Malonek, M.; Szurek, B.; Small, I.; Knoop, V.; Lurin, C. A hypothesis on the identification of the editing enzyme in plant organelles. FEBS Lett. 2007, 581, 4132–4138. [Google Scholar] [CrossRef] [PubMed]
- Rüdinger, M.; Polsakiewicz, M.; Knoop, V. Organellar RNA editing and plant-specific extensions of pentatricopeptide repeat proteins in jungermanniid but not in marchantiid liverworts. Mol. Biol. Evol. 2008, 25, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Knoop, V. When you can’t trust the DNA: RNA editing changes transcript sequences. Cell. Mol. Life Sci. 2011, 68, 567–586. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, M.; Sugita, M. RNA editing and its molecular mechanism in plant organelles. Genes 2016, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Zauner, S.; Grilinger, D.; Laatsch, T.; Kowallik, K.V.; Maier, U.G. Substitutional editing of transcripts from genes of cyanobacterial origin in the dinoflagellate Ceratium horridum. FEBS Lett. 2004, 577, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Adl, S.M.; Simpson, A.G.B.; Lane, C.E.; Lues, J.; Bass, D.; Bowser, S.S.; Brown, M.W.; Burk, F.; Dunthorn, M.; Hampl, V.; et al. The Revised Classification of Eukaryotes. J. Eukaryot. Microbiol. 2012, 59, 429–514. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J. The endosymbiotic origin, diversification and fate of plastids. Phil. Trans. R. Soc. B 2010, 365, 729–748. [Google Scholar] [CrossRef] [PubMed]
- Dorrell, R.G.; Howe, C.J. Integration of plastids with their hosts: Lessons learned from dinoflagellates. Proc. Natl. Acad. Sci. USA 2015, 112, 10247–10254. [Google Scholar] [CrossRef] [PubMed]
- Gagat, P.; Bodyl, A.; Mackiewicz, P.; Stiller, J.W. Tertiary plastid endosymbiosis in dinoflagellates. In Endosymbiosis; Loffelhardt, W., Ed.; Springer: Vienna, Austria, 2014; pp. 233–290. [Google Scholar]
- Dang, Y.; Green, B.R. Substitutional editing of Heterocapsa triquetra chloroplast transcripts and a folding model for its divergent 16S rRNA. Gene 2009, 442, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Morse, D. Rampant polyuridylylation of plastid gene transcripts in the dinoflagellate Lingulodinium. Nucleic Acids Res. 2006, 34, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Mungpakdee, S.; Shinzato, C.; Takeuchi, T.; Kawashima, T.; Koyanagi, R.; Hisata, K.; Tanaka, M.; Goto, H.; Fujie, M.; Lin, S.; et al. Massive gene transfer and extensive RNA editing of a symbiotic dinoflagellate plastid genome. Genome Biol. Evol. 2014, 6, 1408–1422. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. PREP-Mt: Predictive RNA editor for plant mitochondrial genes. BMC Bioinform. 2005, 6, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes, and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Li, Y. Prediction of C-to-U RNA editing sites in plant mitochondria using both biochemical and evolutionary information. J. Theoret. Biol. 2008, 253, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Gopal, S. Genetic algorithm learning as a robust approach to RNA editing site prediction. BMC Bioinform. 2006, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Cummings, M.P.; Myers, D.S. Simple statistical models predict C-to-U edited sites in plant mitochondrial RNA. BMC Bioinform. 2004, 5, 132. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.; Knoop, V. PREPACT 2.0: Predicting C-to-U and U-to-C RNA editing in organelle genome sequences with multiple references and curated RNA editing annotation. Bioinform. Biol. Insights 2013, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Karol, K.G.; McCourt, R.M.; Cimino, M.T.; Delwiche, D.F. The closest living relatives of land plants. Science 2001, 294, 2351–2353. [Google Scholar] [CrossRef] [PubMed]
- Leliaert, F.; Smith, D.R.; Moreau, H.; Herron, M.D.; Verbruggen, H.; Delwiche, C.F.; De Clerck, O. Phylogeny and Molecular Evolution of the Green Algae. Crit. Rev. Plant. Sci. 2012, 31, 1–46. [Google Scholar] [CrossRef]
- Harris, E.H. The Chlamydomonas Sourcebook, 2nd ed.; Harris, E.H., Stern, D.B., Witman, G.B., Eds.; Academic Press: Oxford, UK, 2009. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Turmel, M.; Otis, C.; Lemieux, C. The chloroplast genome sequence of Chara vulgaris sheds new light into the closest green algal relatives of land plants. Mol. Biol. Evol. 2006, 23, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Turmel, M.; Otis, C.; Lemieux, C. The mitochondrial genome of Chara vulgaris: Insights into the mitochondrial DNA architecture of the last common ancestor of green algae and land plants. Plant Cell 2003, 15, 1888–1903. [Google Scholar] [CrossRef] [PubMed]
- Chateigner-Boutin, A.L.; Small, I. A rapid high-throughput method for the detection and quantification of RNA editing based on high-resolution melting of amplicons. Nucleic Acids Res. 2007, 35, e114. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Dobosy, J.R.; Rose, S.D.; Beltz, K.R.; Rupp, S.M.; Powers, K.M.; Behlke, M.A.; Walter, J.A. RNase H-dependent PCR (rhPCR): Improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Rieder, L.E.; Savva, Y.A.; Reyna, M.A.; Chang, Y.J.; Dorsky, J.S.; Rezaei, A.; Reenan, R.A. Dynamic response of RNA editing to temperature in Drosophila. BMC Biol. 2015, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.B.; Goldschmidt-Clermont, M.; Hanson, M.R. Chloroplast RNA metabolism. Annu. Rev. Plant. Biol. 2010, 61, 125–155. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Linneweber, C.; Small, I. Pentatricopeptide repeat proteins: A socket set for organelle gene expression. Trends Plant Sci. 2008, 13, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Small, I. The evolution of RNA editing and pentatricopeptide repeat genes. New Phytol. 2011, 191, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Kugita, M.; Yamamoto, Y.; Fujikawa, T.; Matsumoto, T.; Yoshinaga, K. RNA editing in hornwort chloroplast makes more than half the genes functional. Nucleic Acids Res. 2003, 31, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Sugita, M. Tissue- and stage-specific RNA editing of rps14 transcripts in moss (Physcomitrella patens) chloroplasts. J. Plant Physiol. 2004, 161, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, E.; Sugita, M. The moss Physcomitrella patens, a model plant for the study of RNA editing in plant organelles. Plant. Signal. Behav. 2010, 5, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Dorrell, R.G.; Hinksman, G.A.; Howe, C.J. Diversity of transcripts and transcript processing forms in plastids of the dinoflagellate alga Karenia mikimotoiPlant. Mol. Biol. 2016, 90, 233–247. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Chara vulgaris chondriome SNPs compared to NC_005255 | |||
|---|---|---|---|
| SNP location | nt change | gene | |
| 65107 | G → C | rns | |
| 65109 | T → G | rns | |
| 65189 | C → T | rns | |
| 66731 | G → C | rns | |
| 66768 | T → C | rns | |
| 66802 | A → C | rns | |
| Chara vulgaris plastome SNPs compared to NC_008097 | |||
| SNP location | nt change | gene | effect |
| 7191 | T → A | intergenic | |
| 24408 | G → T | intergenic | |
| 37112 | G → T | ycf3 intron 1 | |
| 42310 | T → A | intergenic | |
| 44130 | C → G | intergenic | |
| 44734 | G → A | intergenic | |
| 44967 | T → C | intergenic | |
| 61795 | C → T | trnK(uuu) intron 1 | |
| 87704 | C → G | intergenic | |
| 92194 | G → T | intergenic | |
| 94397 | T → G | intergenic | |
| 100725 | G → A | intergenic | |
| 107118 | G → A | intergenic | |
| 115615 | A → C | petB intron 1 | |
| 126737 | A → T | intergenic | |
| 129523 | G → A | rps3 | synonymous |
| 129668 | C → A | rpl22 | G115V |
| 137046 | T → G | intergenic | |
| 156998 | G → C | intergenic | |
| 157889 | G → C | intergenic | |
| 183703 | A → C | intergenic | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cahoon, A.B.; Nauss, J.A.; Stanley, C.D.; Qureshi, A. Deep Transcriptome Sequencing of Two Green Algae, Chara vulgaris and Chlamydomonas reinhardtii, Provides No Evidence of Organellar RNA Editing. Genes 2017, 8, 80. https://doi.org/10.3390/genes8020080
Cahoon AB, Nauss JA, Stanley CD, Qureshi A. Deep Transcriptome Sequencing of Two Green Algae, Chara vulgaris and Chlamydomonas reinhardtii, Provides No Evidence of Organellar RNA Editing. Genes. 2017; 8(2):80. https://doi.org/10.3390/genes8020080
Chicago/Turabian StyleCahoon, A. Bruce, John A. Nauss, Conner D. Stanley, and Ali Qureshi. 2017. "Deep Transcriptome Sequencing of Two Green Algae, Chara vulgaris and Chlamydomonas reinhardtii, Provides No Evidence of Organellar RNA Editing" Genes 8, no. 2: 80. https://doi.org/10.3390/genes8020080
APA StyleCahoon, A. B., Nauss, J. A., Stanley, C. D., & Qureshi, A. (2017). Deep Transcriptome Sequencing of Two Green Algae, Chara vulgaris and Chlamydomonas reinhardtii, Provides No Evidence of Organellar RNA Editing. Genes, 8(2), 80. https://doi.org/10.3390/genes8020080