
The Diabetes-Linked Transcription Factor PAX4: From Gene to Functional Consequences



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
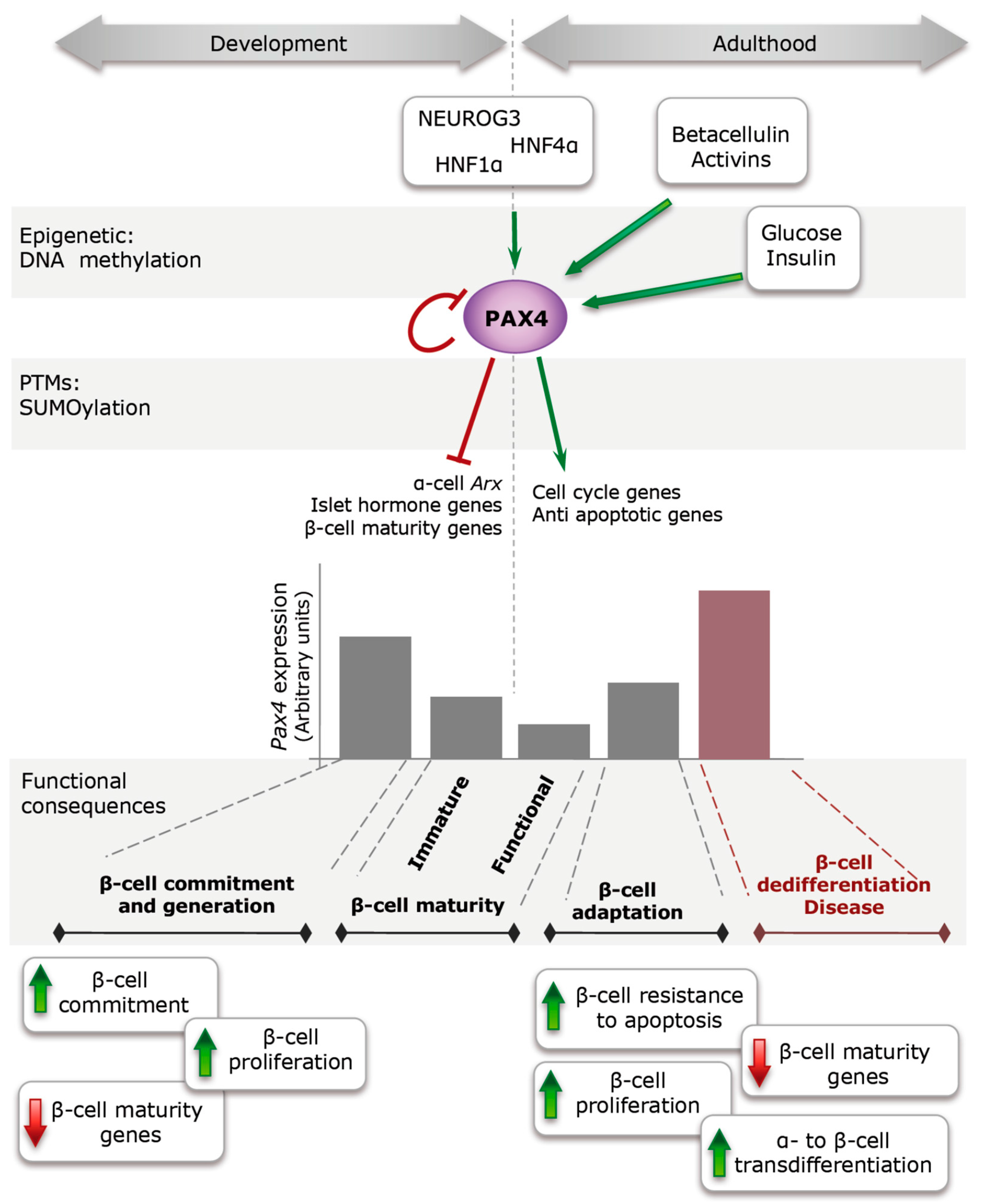
:1. Introduction
2. PAX4 in Islet Physiology: Key Player in β-Cell Generation, Survival and Proliferation
2.1. PAX4 Essential Role for β-Cell Generation during Embryogenesis
2.2. PAX4 Implications in Adult Islet Plasticity
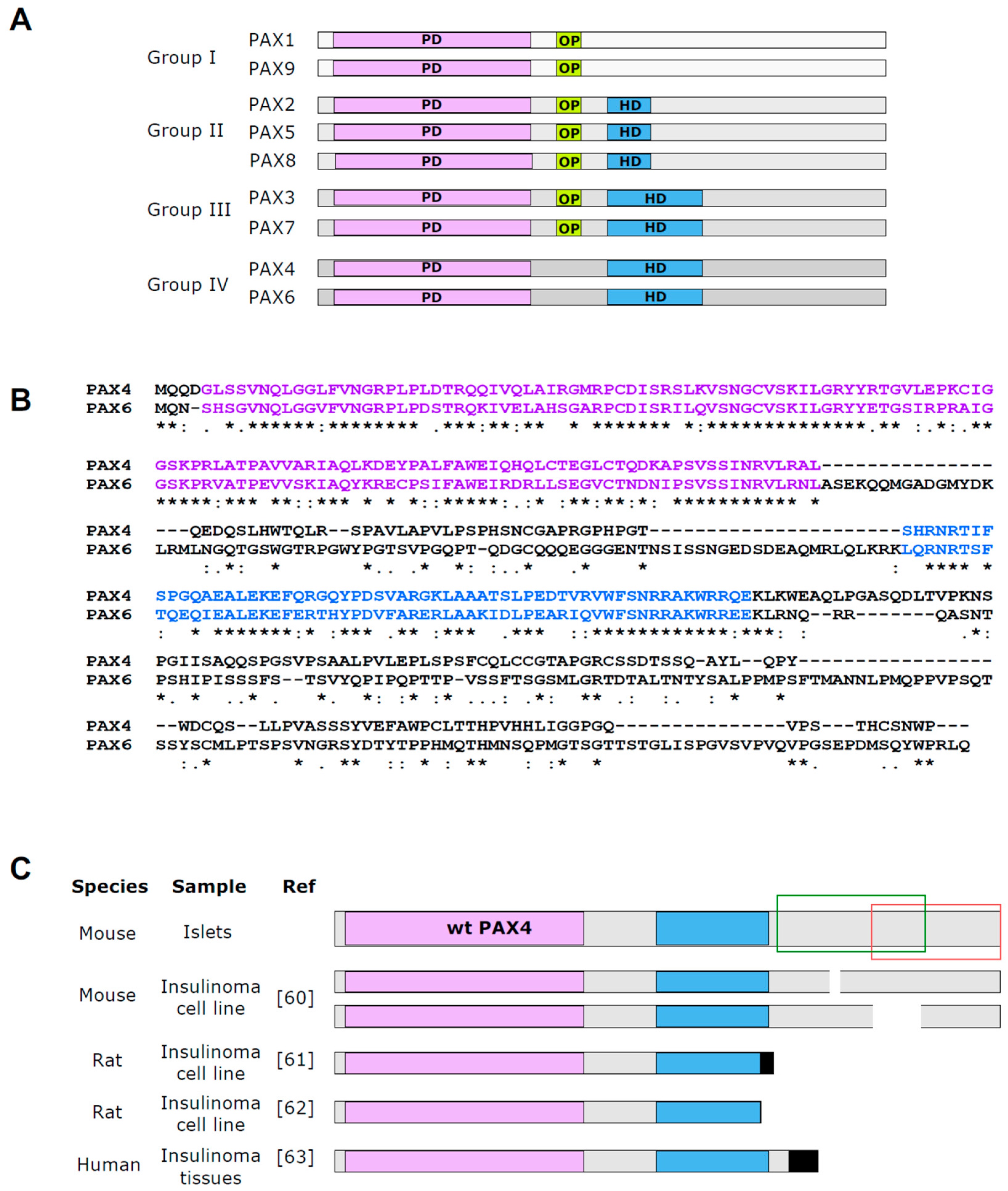
3. PAX4 Molecular Structure and Mechanism of Action
4. PAX4 Mechanism of Action: Downstream Regulated Genes
5. PAX4 Regulation
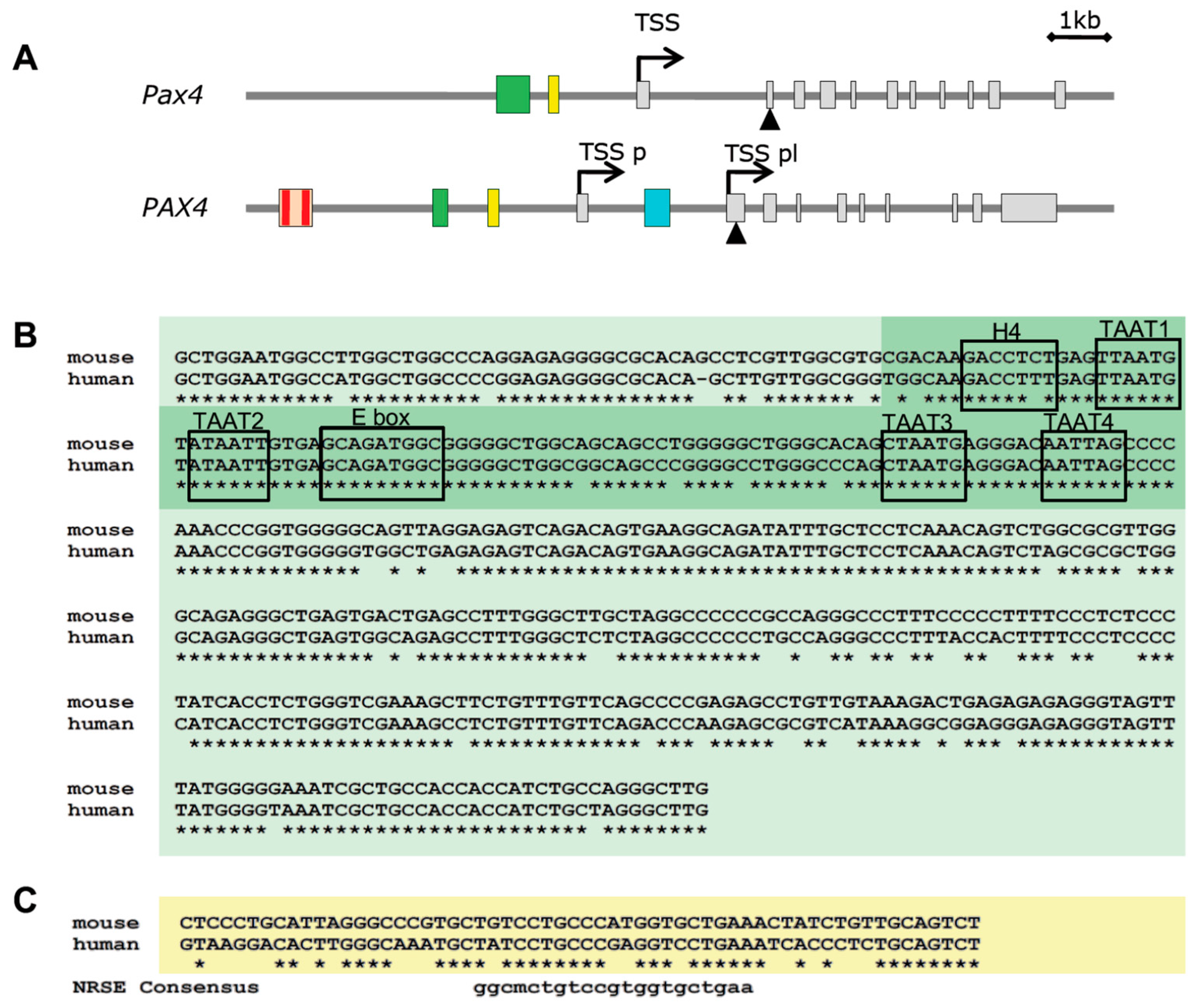
5.1. Epigenetic Regulation of PAX4
5.2. Genetic Regulation of PAX4
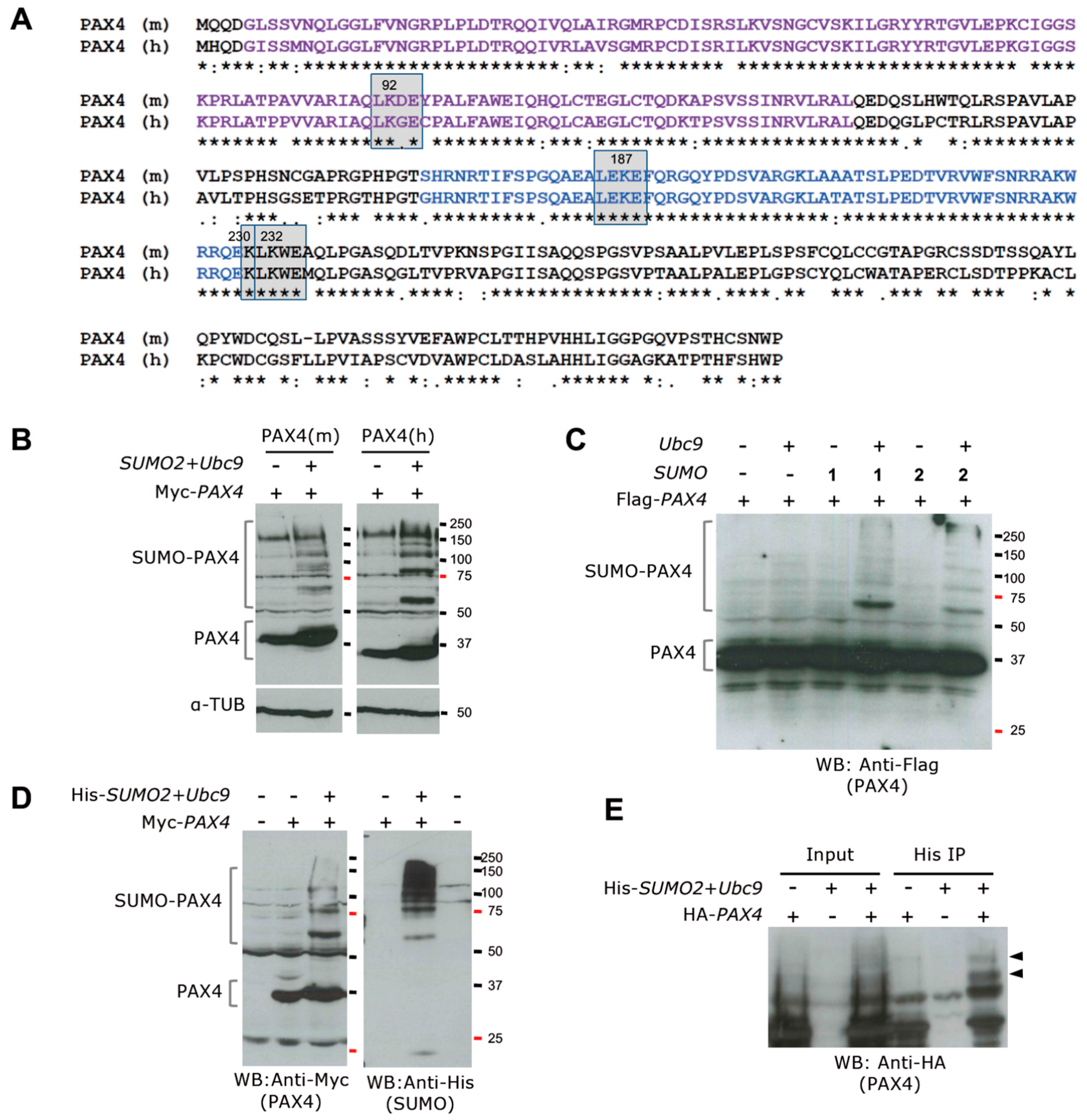
5.3. Posttranslational Regulation of PAX4
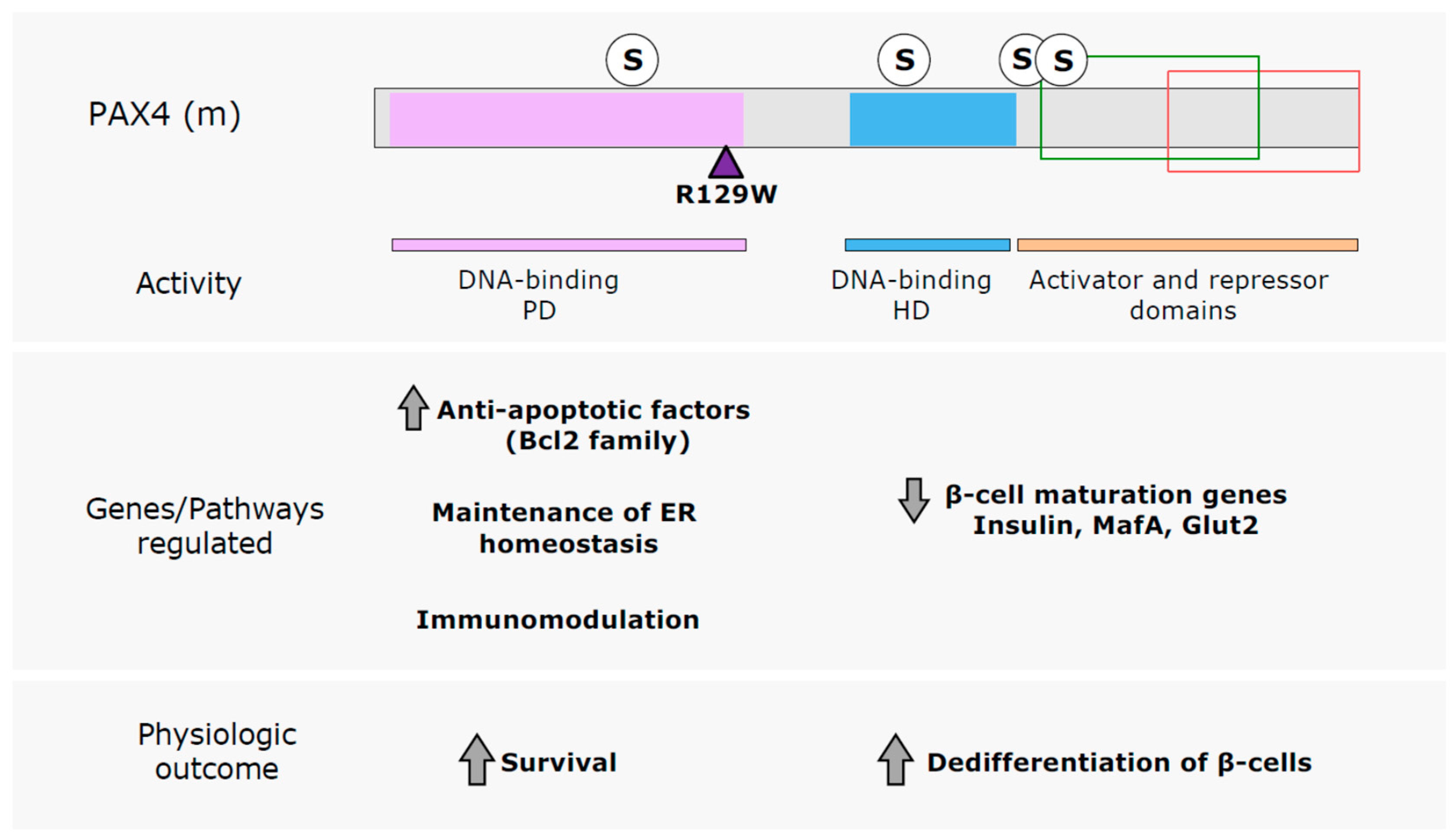
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Association, A.D. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. 1), S81–S90. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Nguyen, X.M.; Lane, J.; Wang, P. Relationship between obesity and diabetes in a US adult population: Findings from the National Health and Nutrition Examination Survey, 1999–2006. Obes. Surg. 2011, 21, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Maeda, S. Genetics of type 2 diabetes: The GWAS era and future perspectives [review]. Endocr. J. 2011, 58, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Boitard, C.; Accili, D.; Ahren, B.; Cerasi, E.; Seino, S.; Thorens, B. The hyperstimulated beta-cell: Prelude to diabetes? Diabetes Obes. Metab. 2012, 14 (Suppl. 3), 4–8. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.; Pociot, F. Genetics of diabetes—Are we missing the genes or the disease? Mol. Cell. Endocrinol. 2014, 382, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Wheeler, B.; Sampson, J.; Hartge, P.; Chanock, S.J.; Park, J.H. Projecting the performance of risk prediction based on polygenic analyses of genome-wide association studies. Nat. Genet. 2013, 45, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Zuk, O.; Hechter, E.; Sunyaev, S.R.; Lander, E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 2012, 109, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Chen, C.H.; Hu, C.; Long, J.; Ong, R.T.; Sim, X.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east asians. Nat. Genet. 2012, 44, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.C.; Hu, C.; Tam, C.H.; Zhang, R.; Kwan, P.; Leung, T.F.; Thomas, G.N.; Go, M.J.; Hara, K.; Sim, X.; et al. Genome-wide association study in a chinese population identifies a susceptibility locus for type 2 diabetes at 7q32 near pax4. Diabetologia 2013, 56, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Lorenzo, P.I.; Lopez-Noriega, L.; Gauthier, B.R. Targeting pancreatic expressed PAX genes for the treatment of diabetes mellitus and pancreatic neuroendocrine tumors. Expert Opin. Ther. Targets 2016, 21, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Sujjitjoon, J.; Kooptiwut, S.; Chongjaroen, N.; Tangjittipokin, W.; Plengvidhya, N.; Yenchitsomanus, P.T. Aberrant mRNA splicing of paired box 4 (PAX4) IVS7–1G>A mutation causing maturity-onset diabetes of the young, type 9. Acta Diabetol. 2016, 53, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Robson, E.J.; He, S.J.; Eccles, M.R. A PANorama of PAX genes in cancer and development. Nat. Rev. Cancer 2006, 6, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Powell, S.K.; Plummer, R.S.; Young, K.P.; Ruggeri, B.A. PAX genes: Roles in development, pathophysiology, and cancer. Biochem. Pharmacol. 2007, 73, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Blake, J.A.; Thomas, M.; Thompson, J.A.; White, R.; Ziman, M. Perplexing Pax: From puzzle to paradigm. Dev. Dyn. 2008, 237, 2791–2803. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fang, W.H.; Krupinski, J.; Kumar, S.; Slevin, M.; Kumar, P. Pax genes in embryogenesis and oncogenesis. J. Cell. Mol. Med. 2008, 12, 2281–2294. [Google Scholar] [CrossRef] [PubMed]
- Blake, J.A.; Ziman, M.R. Pax genes: Regulators of lineage specification and progenitor cell maintenance. Development 2014, 141, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Pineda, B. The gene Pax4 is an essential regulator of pancreatic beta-cell development. Mol. Cells 2004, 18, 289–294. [Google Scholar] [PubMed]
- Brun, T.; Gauthier, B.R. A focus on the role of Pax4 in mature pancreatic islet β-cell expansion and survival in health and disease. J. Mol. Endocrinol. 2008, 40, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, T.; Avolio, F.; Courtney, M.; Vieira, A.; Druelle, N.; Ben-Othman, N.; Hadzic, B.; Navarro, S.; Collombat, P. Pax4 acts as a key player in pancreas development and plasticity. Semin. Cell Dev. Biol. 2015, 44, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, A.L.; Li, S.; Jones, K.; Melton, D.A. Notch signaling reveals developmental plasticity of Pax4(+) pancreatic endocrine progenitors and shunts them to a duct fate. Mech. Dev. 2007, 124, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, P.I.; Fuente-Martin, E.; Brun, T.; Cobo-Vuilleumier, N.; Jimenez-Moreno, C.M.; Irene, G.H.G.; Lopez Noriega, L.; Mellado-Gil, J.M.; Martin-Montalvo, A.; Soria, B.; et al. Pax4 defines an expandable beta-cell subpopulation in the adult pancreatic islet. Sci. Rep. 2015, 5, 15672. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Pineda, B.; Chowdhury, K.; Torres, M.; Oliver, G.; Gruss, P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 1997, 386, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Elghazi, L.; Parker, S.E.; Kizilocak, H.; Asano, M.; Sussel, L.; Sosa-Pineda, B. The concerted activities of Pax4 and Nkx2.2 are essential to initiate pancreatic beta-cell differentiation. Dev. Biol. 2004, 266, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Collombat, P.; Hecksher-Sorensen, J.; Broccoli, V.; Krull, J.; Ponte, I.; Mundiger, T.; Smith, J.; Gruss, P.; Serup, P.; Mansouri, A. The simultaneous loss of Arx and Pax4 genes promotes a somatostatin-producing cell fate specification at the expense of the alpha- and beta-cell lineages in the mouse endocrine pancreas. Development 2005, 132, 2969–2980. [Google Scholar] [CrossRef] [PubMed]
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009, 138, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Blyszczuk, P.; Czyz, J.; Kania, G.; Wagner, M.; Roll, U.; St-Onge, L.; Wobus, A.M. Expression of Pax4 in embryonic stem cells promotes differentiation of nestin-positive progenitor and insulin-producing cells. Proc. Natl. Acad. Sci. USA 2003, 100, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.T.; Kao, C.L.; Lee, K.H.; Chang, Y.L.; Chiou, S.H.; Tsai, F.T.; Tsai, T.H.; Sheu, D.C.; Ho, L.L.; Ku, H.H. Enhancement of insulin-producing cell differentiation from embryonic stem cells using pax4-nucleofection method. World J. Gastroenterol. 2007, 13, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Liew, C.G.; Shah, N.N.; Briston, S.J.; Shepherd, R.M.; Khoo, C.P.; Dunne, M.J.; Moore, H.D.; Cosgrove, K.E.; Andrews, P.W. PAX4 enhances beta-cell differentiation of human embryonic stem cells. PLoS ONE 2008, 3, e1783. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.J.; Docherty, H.M.; Chen, Y.; Docherty, K. Efficient differentiation of AR42J cells towards insulin-producing cells using pancreatic transcription factors in combination with growth factors. Mol. Cell. Endocrinol. 2012, 358, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Berneman-Zeitouni, D.; Molakandov, K.; Elgart, M.; Mor, E.; Fornoni, A.; Dominguez, M.R.; Kerr-Conte, J.; Ott, M.; Meivar-Levy, I.; Ferber, S. The temporal and hierarchical control of transcription factors-induced liver to pancreas transdifferentiation. PLoS ONE 2014, 9, e87812. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.K.; Baker, R.K.; Kieffer, T.J. Overexpression of PAX4 reduces glucagon expression in differentiating hESCs. Islets 2014, 6, e29236. [Google Scholar] [CrossRef] [PubMed]
- Soria, B.; Gauthier, B.R.; Martin, F.; Tejedo, J.R.; Bedoya, F.J.; Rojas, A.; Hmadcha, A. Using stem cells to produce insulin. Expert Opin. Biol. Ther. 2015, 15, 1469–1489. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; Franklin, I.; St-Onge, L.; Biason-Lauber, A.; Schoenle, E.; Wollheim, C.B.; Gauthier, B.R. The diabetes-linked transcription factor Pax4 promotes beta-cell proliferation and survival in rat and human islets. J. Cell Biol. 2004, 167, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; He, K.H.; Lupi, R.; Boehm, B.; Wojtusciszyn, A.; Sauter, N.; Donath, M.; Marchetti, P.; Maedler, K.; Gauthier, B.R. The diabetes-linked transcription factor Pax4 is expressed in human pancreatic islets and is activated by mitogens and GLP-1. Hum. Mol. Genet. 2008, 17, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, G.; Lan, M.S.; Zhang, S.; Fan, W.; Wang, H.; Lu, D. Pax4 paired domain mediates direct protein transduction into mammalian cells. Endocrinology 2007, 148, 5558–5565. [Google Scholar] [CrossRef] [PubMed]
- Rezende, L.F.; Stoppiglia, L.F.; Souza, K.L.; Negro, A.; Langone, F.; Boschero, A.C. Ciliary neurotrophic factor promotes survival of neonatal rat islets via the BCL-2 anti-apoptotic pathway. J. Endocrinol. 2007, 195, 157–165. [Google Scholar] [CrossRef] [PubMed]
- He, K.H.H.; Lorenzo, P.I.; Brun, T.; Jimenez Moreno, C.M.; Aeberhard, D.; Ortega, J.V.; Cornu, M.; Thorel, F.; Gjinovci, A.; Thorens, B.; et al. In Vivo conditional Pax4 overexpression in mature islet {beta}-cells prevents stress-induced hyperglycemia in mice. Diabetes 2011, 60, 1705–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, T.; Duhamel, D.L.; He, K.H.H.; Wollheim, C.B.; Gauthier, B.R. The transcription factor Pax4 acts as a survival gene in the insulinoma INS1E cells. Oncogene 2007, 26, 4261–4271. [Google Scholar] [CrossRef] [PubMed]
- Mellado-Gil, J.M.; Jimenez-Moreno, C.M.; Martin-Montalvo, A.; Alvarez-Mercado, A.I.; Fuente-Martin, E.; Cobo-Vuilleumier, N.; Lorenzo, P.I.; Bru-Tari, E.; de Gracia Herrera-Gomez, I.; Lopez-Noriega, L.; et al. PAX4 preserves endoplasmic reticulum integrity preventing beta cell degeneration in a mouse model of type 1 diabetes mellitus. Diabetologia 2016, 59, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Ripoche, D.; Charbord, J.; Hennino, A.; Teinturier, R.; Bonnavion, R.; Jaafar, R.; Goehrig, D.; Cordier-Bussat, M.; Ritvos, O.; Zhang, C.X.; et al. ActivinB is induced in insulinoma to promote tumor plasticity through a beta-cell-induced dedifferentiation. Mol. Cell. Biol. 2016, 36, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Al-Hasani, K.; Pfeifer, A.; Courtney, M.; Ben-Othman, N.; Gjernes, E.; Vieira, A.; Druelle, N.; Avolio, F.; Ravassard, P.; Leuckx, G.; et al. Adult duct-lining cells can reprogram into beta-like cells able to counter repeated cycles of toxin-induced diabetes. Dev. Cell 2013, 26, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fava, G.E.; Wang, H.; Mauvais-Jarvis, F.; Fonseca, V.A.; Wu, H. Pax4 gene transfer induces α-to-β cell phenotypic conversion and confers therapeutic benefits for diabetes treatment. Mol. Ther. 2016, 24, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Czerny, T.; Busslinger, M. DNA-binding and transactivation properties of Pax-6: Three amino acids in the paired domain are responsible for the different sequence recognition of Pax-6 and BSAP (Pax-5). Mol. Cell. Biol. 1995, 15, 2858–2871. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.B.; Ee, H.C.; Conners, J.R.; German, M.S. Paired-homeodomain transcription factor PAX4 acts as a transcriptional repressor in early pancreatic development. Mol. Cell. Biol. 1999, 19, 8272–8280. [Google Scholar] [CrossRef] [PubMed]
- Czerny, T.; Schaffner, G.; Busslinger, M. DNA sequence recognition by Pax proteins: Bipartite structure of the paired domain and its binding site. Genes Dev. 1993, 7, 2048–2061. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.A.; Glaser, T.; Cai, J.; Jepeal, L.; Walton, D.S.; Maas, R.L. Two independent and interactive DNA-binding subdomains of the Pax6 paired domain are regulated by alternative splicing. Genes Dev. 1994, 8, 2022–2034. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.E.; Rould, M.A.; Xu, W.; Epstein, J.A.; Maas, R.L.; Pabo, C.O. Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev. 1999, 13, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Apuzzo, S.; Abdelhakim, A.; Fortin, A.S.; Gros, P. Cross-talk between the paired domain and the homeodomain of Pax3: DNA binding by each domain causes a structural change in the other domain, supporting interdependence for DNA binding. J. Biol. Chem. 2004, 279, 33601–33612. [Google Scholar] [CrossRef] [PubMed]
- Mayran, A.; Pelletier, A.; Drouin, J. Pax factors in transcription and epigenetic remodelling. Semin. Cell Dev. Biol. 2015, 44, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.; Desplan, C. Cooperative interactions between paired domain and homeodomain. Development 1996, 122, 2639–2650. [Google Scholar] [PubMed]
- Singh, S.; Stellrecht, C.M.; Tang, H.K.; Saunders, G.F. Modulation of PAX6 homeodomain function by the paired domain. J. Biol. Chem. 2000, 275, 17306–17313. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Gorlov, I.P.; Chao, L.Y.; Singh, S.; Saunders, G.F. PAX6, paired domain influences sequence recognition by the homeodomain. J. Biol. Chem. 2002, 277, 49488–49494. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Gong, L.; Deng, M.; Zhang, L.; Sun, S.; Liu, J.; Ma, H.; Yuan, D.; Chen, P.C.; Hu, X.; et al. Sumoylation activates the transcriptional activity of Pax-6, an important transcription factor for eye and brain development. Proc. Natl. Acad. Sci. USA 2010, 107, 21034–21039. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, Y.; Kajimoto, Y.; Yasuda, T.; Matsuoka, T.A.; Kaneto, H.; Umayahara, Y.; Fujita, N.; Watada, H.; Miyazaki, J.I.; Yamasaki, Y.; et al. Identification of a portable repression domain and an E1A-responsive activation domain in Pax4: A possible role of Pax4 as a transcriptional repressor in the pancreas. Mol. Cell. Biol. 1999, 19, 8281–8291. [Google Scholar] [CrossRef] [PubMed]
- Ritz-Laser, B.; Estreicher, A.; Gauthier, B.R.; Mamin, A.; Edlund, H.; Philippe, J. The pancreatic beta-cell-specific transcription factor Pax-4 inhibits glucagon gene expression through Pax-6. Diabetologia 2002, 45, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Shimajiri, Y.; Sanke, T.; Furuta, H.; Hanabusa, T.; Nakagawa, T.; Fujitani, Y.; Kajimoto, Y.; Takasu, N.; Nanjo, K. A missense mutation of Pax4 gene (R121W) is associated with type 2 diabetes in Japanese. Diabetes 2001, 50, 2864–2869. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, Y.; Uchikawa, M.; Tanouchi, A.; Sekido, R.; Kondoh, H. Pax6 and SOX2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes Dev. 2001, 15, 1272–1286. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Epstein, J.A. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Hum. Mol. Genet. 2003, 12, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Jepeal, L.; Edwards, J.G.; Young, S.R.; Favor, J.; Maas, R.L. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat. Genet. 1994, 7, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Nomiyama, J.; Nakai, K.; Matsutani, A.; Tanizawa, Y.; Oka, Y. Isolation of full-length cDNA of mouse PAX4 gene and identification of its human homologue. Biochem. Biophys. Res. Commun. 1998, 243, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, Y.; Yagui, K.; Sakurai, K.; Hashimoto, N.; Saito, Y.; Kanatsuka, A. Molecular cloning of rat Pax4: Identification of four isoforms in rat insulinoma cells. Biochem. Biophys. Res. Commun. 1998, 248, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.C.; Cragg, H.; Elrick, L.J.; Macfarlane, W.M.; Shennan, K.I.; Docherty, K. Inhibitory effect of Pax4 on the human insulin and islet amyloid polypeptide (IAPP) promoters. FEBS Lett. 1999, 463, 53–57. [Google Scholar] [CrossRef]
- Miyamoto, T.; Kakizawa, T.; Ichikawa, K.; Nishio, S.; Kajikawa, S.; Hashizume, K. Expression of dominant negative form of PAX4 in human insulinoma. Biochem. Biophys. Res. Commun. 2001, 282, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Petersen, H.V.; Jorgensen, M.C.; Andersen, F.G.; Jensen, J.; Tove, F.N.; Jorgensen, R.; Madsen, O.D.; Serup, P. Pax4 represses pancreatic glucagon gene expression. Mol. Cell Biol. Res. Commun. 2000, 3, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Elghazi, L.; Martin, S.; Martins, I.; Srinivasan, R.S.; Geng, X.; Sleeman, M.; Collombat, P.; Houghton, J.; Sosa-Pineda, B. Ghrelin is a novel target of Pax4 in endocrine progenitors of the pancreas and duodenum. Dev. Dyn. 2008, 237, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Andersen, F.G.; Jensen, J.; Heller, R.S.; Petersen, H.V.; Larsson, L.I.; Madsen, O.D.; Serup, P. Pax6 and Pdx1 form a functional complex on the rat somatostatin gene upstream enhancer. FEBS Lett. 1999, 445, 315–320. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Koh, A.; El Khattabi, I.; Li, W.C.; Toschi, E.; Jermendy, A.; Juhl, K.; Mao, K.; Weir, G.C.; Sharma, A.; et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia 2011, 54, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Raum, J.C.; Gerrish, K.; Artner, I.; Henderson, E.; Guo, M.; Sussel, L.; Schisler, J.C.; Newgard, C.B.; Stein, R. Foxa2, Nkx2.2, and PDX-1 regulate islet beta-cell-specific mafA expression through conserved sequences located between base pairs -8118 and -7750 upstream from the transcription start site. Mol. Cell. Biol. 2006, 26, 5735–5743. [Google Scholar] [CrossRef] [PubMed]
- Raum, J.C.; Hunter, C.S.; Artner, I.; Henderson, E.; Guo, M.; Elghazi, L.; Sosa-Pineda, B.; Ogihara, T.; Mirmira, R.G.; Sussel, L.; et al. Islet beta-cell-specific MafA transcription requires the 5'-flanking conserved region 3 control domain. Mol. Cell. Biol. 2010, 30, 4234–4244. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.C.; Shieh, S.J.; Sytwu, H.K. Attenuation of Th1 response through galectin-9 and T-cell Ig mucin 3 interaction inhibits autoimmune diabetes in NOD mice. Eur. J. Immunol. 2009, 39, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, M.; Wada, J.; Sugiyama, K.; Nakatsuka, A.; Teshigawara, S.; Murakami, K.; Inoue, K.; Terami, T.; Katayama, A.; Eguchi, J.; et al. Galectin-9 and T cell immunoglobulin mucin-3 pathway is a therapeutic target for type 1 diabetes. Endocrinology 2012, 153, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.C.; Kuo, C.C.; Wang, Y.L.; Lin, M.H.; Linju Yen, B.; Chang, D.M.; Sytwu, H.K. Overexpression of galectin-9 in islets prolongs grafts survival via downregulation of Th1 responses. Cell Transpl. 2013, 22, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Nagai, H.; Ohno, T.; Ohashi, H.; Murohara, T.; Saito, H.; Kinoshita, T. Aberrant DNA demethylation in promoter region and aberrant expression of mRNA of PAX4 gene in hematologic malignancies. Leuk. Res. 2006, 30, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Flosseau, K.; Ma, F.X.; Blondeau, B.; Mai, A.; Bassel-Duby, R.; Ravassard, P.; Olson, E.N.; Haumaitre, C.; Scharfmann, R. Specific control of pancreatic endocrine beta- and delta-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 2011, 60, 2861–2871. [Google Scholar] [CrossRef] [PubMed]
- Brink, C.; Chowdhury, K.; Gruss, P. Pax4 regulatory elements mediate beta cell specific expression in the pancreas. Mech. Dev. 2001, 100, 37–43. [Google Scholar] [CrossRef]
- Brink, C.; Gruss, P. DNA sequence motifs conserved in endocrine promoters are essential for Pax4 expression. Dev. Dyn. 2003, 228, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.B.; Watada, H.; Scheel, D.W.; Mrejen, C.; German, M.S. Autoregulation and maturity onset diabetes of the young transcription factors control the human PAX4 promoter. J. Biol. Chem. 2000, 275, 36910–36919. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.B.; Gasa, R.; Watada, H.; Wang, J.; Griffen, S.C.; German, M.S. Neurogenin3 and hepatic nuclear factor 1 cooperate in activating pancreatic expression of Pax4. J. Biol. Chem. 2003, 278, 38254–38259. [Google Scholar] [CrossRef] [PubMed]
- Heremans, Y.; Van De Casteele, M.; in’t Veld, P.; Gradwohl, G.; Serup, P.; Madsen, O.; Pipeleers, D.; Heimberg, H. Recapitulation of embryonic neuroendocrine differentiation in adult human pancreatic duct cells expressing neurogenin 3. J. Cell Biol. 2002, 159, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Kemp, D.M.; Lin, J.C.; Habener, J.F. Regulation of Pax4 paired homeodomain gene by neuron-restrictive silencer factor. J. Biol. Chem. 2003, 278, 35057–35062. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Allagnat, F.; Chaffard, G.; Caille, D.; Fukuda, M.; Regazzi, R.; Abderrahmani, A.; Waeber, G.; Meda, P.; Maechler, P.; et al. Functional significance of repressor element 1 silencing transcription factor (REST) target genes in pancreatic beta cells. Diabetologia 2008, 51, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Kim, Y.H.; Sever, D.; Mao, C.A.; Haefliger, J.A.; Grapin-Botton, A. REST represses a subset of the pancreatic endocrine differentiation program. Dev. Biol. 2015, 405, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Mellado-Gil, J.M.; Fuente-Martin, E.; Lorenzo, P.I.; Bermudez-Silva, F.J.; Rojo-Martinez, G.; Romero-Zerbo, S.Y.; Campos-Caro, A.; Aguilar-Diosdado, M.; Gauthier, B.R. The diabetes-link factor HMG20A maintains islet beta cell metabolic maturity. Diabetologia 2016, 59, S194. [Google Scholar]
- Huotari, M.A.; Miettinen, P.J.; Palgi, J.; Koivisto, T.; Ustinov, J.; Harari, D.; Yarden, Y.; Otonkoski, T. ErbB signaling regulates lineage determination of developing pancreatic islet cells in embryonic organ culture. Endocrinology 2002, 143, 4437–4446. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, S.; Van Antwerp, M.E.; Iniguez-Lluhi, J.A. Direct and distinguishable inhibitory roles for SUMO isoforms in the control of transcriptional synergy. Proc. Natl. Acad. Sci. USA 2003, 100, 15758–15763. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Mizusaki, H.; Mukai, T.; Ogawa, H.; Baba, D.; Shirakawa, M.; Hatakeyama, S.; Nakayama, K.I.; Yamamoto, H.; Kikuchi, A.; et al. Small ubiquitin-like modifier 1 (SUMO-1) modification of the synergy control motif of Ad4 binding protein/steroidogenic factor 1 (Ad4BP/SF-1) regulates synergistic transcription between AD4BP/SF-1 and Sox9. Mol. Endocrinol. 2004, 18, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Chupreta, S.; Brevig, H.; Bai, L.; Merchant, J.L.; Iniguez-Lluhi, J.A. Sumoylation-dependent control of homotypic and heterotypic synergy by the Kruppel-type zinc finger protein ZBP-89. J. Biol. Chem. 2007, 282, 36155–36166. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dominguez, M.; Reyes, J.C. SUMO association with repressor complexes, emerging routes for transcriptional control. Biochim. Biophys. Acta 2009, 1789, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Molvaersmyr, A.K.; Saether, T.; Gilfillan, S.; Lorenzo, P.I.; Kvaloy, H.; Matre, V.; Gabrielsen, O.S. A SUMO-regulated activation function controls synergy of c-Myb through a repressor-activator switch leading to differential p300 recruitment. Nucleic Acids Res. 2010, 38, 4970–4984. [Google Scholar] [CrossRef] [PubMed]
- Alm-Kristiansen, A.H.; Lorenzo, P.I.; Molvaersmyr, A.K.; Matre, V.; Ledsaak, M.; Saether, T.; Gabrielsen, O.S. PIAS1 interacts with FLASH and enhances its co-activation of c-Myb. Mol. Cancer 2011, 10, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sireesh, D.; Bhakkiyalakshmi, E.; Ramkumar, K.M.; Rathinakumar, S.; Jennifer, P.S.; Rajaguru, P.; Paulmurugan, R. Targeting SUMOylation cascade for diabetes management. Curr. Drug Targets 2014, 15, 1094–1106. [Google Scholar] [CrossRef] [PubMed]
- Aribi, M. Candidate genes implicated in type 1 diabetes susceptibility. Curr. Diabetes Rev. 2008, 4, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Hajmrle, C.; Ferdaoussi, M.; Plummer, G.; Spigelman, A.F.; Lai, K.; Manning Fox, J.E.; MacDonald, P.E. SUMOylation protects against IL-1beta-induced apoptosis in INS-1 832/13 cells and human islets. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E664–E673. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, W.; Jiang, W.; Sun, X.; Han, Y.; Ding, M.; Shi, Y.; Deng, H. P21cip-overexpression in the mouse beta cells leads to the improved recovery from streptozotocin-induced diabetes. PLoS ONE 2009, 4, e8344. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Jafarzade, B.S.; Mardani, G. In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides 2017, 87, 50–63. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenzo, P.I.; Juárez-Vicente, F.; Cobo-Vuilleumier, N.; García-Domínguez, M.; Gauthier, B.R. The Diabetes-Linked Transcription Factor PAX4: From Gene to Functional Consequences. Genes 2017, 8, 101. https://doi.org/10.3390/genes8030101
Lorenzo PI, Juárez-Vicente F, Cobo-Vuilleumier N, García-Domínguez M, Gauthier BR. The Diabetes-Linked Transcription Factor PAX4: From Gene to Functional Consequences. Genes. 2017; 8(3):101. https://doi.org/10.3390/genes8030101
Chicago/Turabian StyleLorenzo, Petra I., Francisco Juárez-Vicente, Nadia Cobo-Vuilleumier, Mario García-Domínguez, and Benoit R. Gauthier. 2017. "The Diabetes-Linked Transcription Factor PAX4: From Gene to Functional Consequences" Genes 8, no. 3: 101. https://doi.org/10.3390/genes8030101
APA StyleLorenzo, P. I., Juárez-Vicente, F., Cobo-Vuilleumier, N., García-Domínguez, M., & Gauthier, B. R. (2017). The Diabetes-Linked Transcription Factor PAX4: From Gene to Functional Consequences. Genes, 8(3), 101. https://doi.org/10.3390/genes8030101