
Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling


{kind=link}
Abstract
:1. MYC: A Potent Driver of Cell and Tissue Growth
2. MYC Functional Redundancy Depends on Transcriptional Patterning
3. Patterning MYC Transcription during Animal Development
4. Developmental Signals Patterning MYC Expression
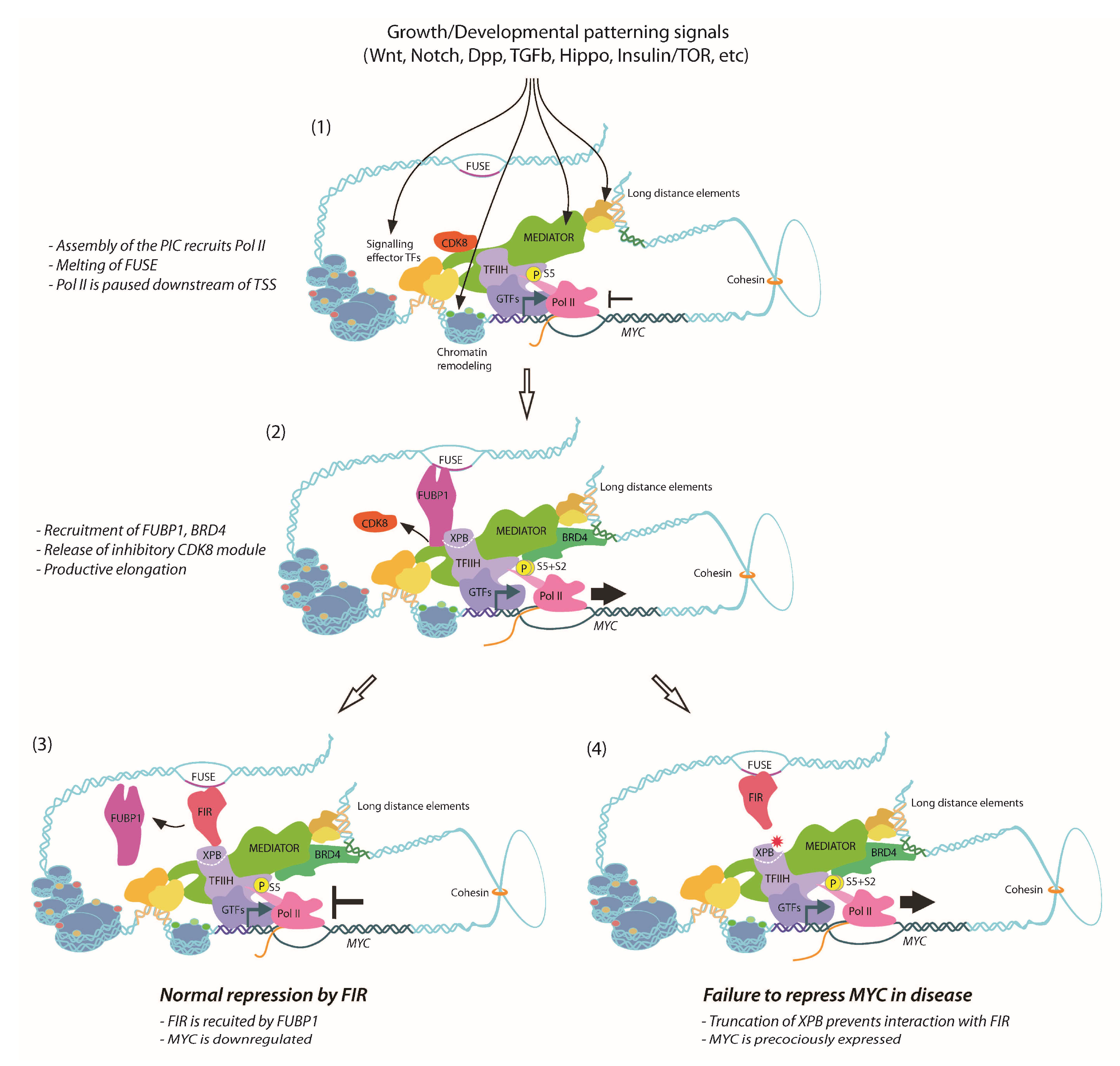
5. MYC Promoter Architecture Reflects Signaling Inputs
6. FUBP1—A Hyperactivator of MYC Transcription
7. Developmental Function of FUBP1
8. FUBP-Interacting Repressor (FIR)—the FUBP1 Antagonist Keeps MYC Quiet
9. Dynamics of FUBP1, FIR and TFIIH Activity on the MYC Promoter
10. Defective FIR-Dependent Repression of MYC in XPB-Related Disease
11. Defective MYC Repression and Tissue Overgrowth in Drosophila Models of XPB-Related Disease
12. Drosophila FUBP1/Psi Interacts with Mediator to Control MYC Transcription
13. MYC Control at A Distance—3D Genomic Architecture in MYC Regulation
14. Targeting MYC for Cancer Treatment
15. Concluding Remarks and Future Considerations
Conflicts of Interest
References
- Levens, D. You Don’t Muck with MYC. Genes Cancer 2010, 1, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harbor Perspect. Med. 2014, 4, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Poortinga, G.; Wall, M.; Sanij, E.; Siwicki, K.; Ellul, J.; Brown, D.; Holloway, T.P.; Hannan, R.D.; McArthur, G.A. c-MYC coordinately regulates ribosomal gene chromatin remodeling and Pol I availability during granulocyte differentiation. Nucleic Acids Res. 2011, 39, 3267–3281. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.S.; Li, L.; Orian, A.; Eisenman, R.N.; Edgar, B.A. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat. Cell Biol. 2005, 7, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Poortinga, G.; Hannan, K.M.; Snelling, H.; Walkley, C.R.; Jenkins, A.; Sharkey, K.; Wall, M.; Brandenburger, Y.; Palatsides, M.; Pearson, R.B.; et al. MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J. 2004, 23, 3325–3335. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Hanson, K.D.; Sedivy, J.M. Effects of c-myc expression on proliferation, quiescence, and the G0 to G1 transition in nontransformed cells. Cell Growth Differ. 1993, 4, 93–104. [Google Scholar] [PubMed]
- Gowda, S.D.; Koler, R.D.; Bagby, G.C. Regulation of C-myc expression during growth and differentiation of normal and leukemic human myeloid progenitor cells. J. Clin. Investig. 1986, 77, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.I.; Lin, Y.; Calame, K. Repression of c-myc is necessary but not sufficient for terminal differentiation of B lymphocytes in vitro. Mol. Cell. Biol. 2000, 20, 8684–8695. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Yu, X.; Zhang, Y.; DeWille, J.W. Myc interacts with Max and Miz1 to repress C/EBPdelta promoter activity and gene expression. Mol. Cancer 2010, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Herkert, B.; Eilers, M. Transcriptional repression: The dark side of myc. Genes Cancer 2010, 1, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Orian, A. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003, 17, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; Grewal, S.S.; Knoepfler, P.S.; Edgar, B.A.; Parkhurst, S.M.; Eisenman, R.N. Genomic binding and transcriptional regulation by the Drosophila Myc and Mnt transcription factors. Cold Spring Harbor Symp. Quant. Biol. 2005, 70, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wu, G.; Zhan, X.; Nolan, A.; Koh, C.; De Marzo, A.; Doan, H.M.; Fan, J.; Cheadle, C.; Fallahi, M.; et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 2011, 6, e26057. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Ziff, E.B. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science 1991, 251, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Sabo, A.; Amati, B. Genome Recognition by MYC. Cold Spring Harbor Perspect. Med. 2014, 4, a014191. [Google Scholar] [CrossRef] [PubMed]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kress, T.R.; Sabò, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Lorenzin, F.; Benary, U.; Baluapuri, A.; Walz, S.; Jung, L.A.; von Eyss, B.; Kisker, C.; Wolf, J.; Eilers, M.; Wolf, E. Different promoter affinities account for specificity in MYC-dependent gene regulation. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Lin, C.Y.; Eilers, M.; Levens, D.L. Taming of the beast: Shaping Myc-dependent amplification. Trends Cell Biol. 2015, 25, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Mathsyaraja, H.; Eisenman, R.N. Parsing Myc Paralogs in Oncogenesis. Cancer Cell 2016, 29, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.C.; Adolphe, C.; Ehninger, A.; Wang, R.A.; Robertson, E.J.; Trumpp, A. Placental rescue reveals a sole requirement for c-Myc in embryonic erythroblast survival and hematopoietic stem cell function. Development 2008, 135, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Malynn, B.A.; de Alboran, I.M.; O'Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar] [PubMed]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E. Differential expression of myc family genes during murine development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Knoepfler, P.S.; Kenney, A.M.; Rowitch, D.H.; de Alborán, I.M.; Olson, J.M.; Eisenman, R.N. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 2006, 66, 8655–8661. [Google Scholar] [CrossRef] [PubMed]
- Vo, B.T.; Wolf, E.; Kawauchi, D.; Gebhardt, A.; Rehg, J.E.; Finkelstein, D.; Walz, S.; Murphy, B.L.; Youn, Y.H.; Han, Y.-G.; et al. The Interaction of Myc with Miz1 Defines Medulloblastoma Subgroup Identity. Cancer Cell 2016, 29, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Benassayag, C.; Montero, L.; Colombie, N.; Gallant, P.; Cribbs, D.; Morello, D. Human c-Myc Isoforms Differentially Regulate Cell Growth and Apoptosis in Drosophila melanogaster. Mol. Cell. Biol. 2005, 25, 9897–9909. [Google Scholar] [CrossRef] [PubMed]
- Schreiber-Agus, N.; Stein, D.; Chen, K.; Goltz, J.S.; Stevens, L.; DePinho, R.A. Drosophila Myc is oncogenic in mammalian cells and plays a role in the diminutive phenotype. Proc. Natl. Acad. Sci. USA 1997, 94, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A.; Prober, D.A.; Edgar, B.A.; Eisenman, R.N.; Gallant, P. Drosophila myc regulates cellular growth during development. Cell 1999, 98, 779–790. [Google Scholar] [CrossRef]
- Prober, D.A.; Edgar, B.A. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 2002, 16, 2286–2299. [Google Scholar] [CrossRef] [PubMed]
- Er Amanda Lee, J.; May Parsons, L.; M Quinn, L. MYC function and regulation in flies: How Drosophila has enlightened MYC cancer biology. AIMS Genet. 2014, 1, 81–98. [Google Scholar] [CrossRef]
- Grifoni, D.; Bellosta, P. Drosophila Myc: A master regulator of cellular performance. Biochim. Biophys. Acta 2015, 1849, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sung, E.; Donlin-Asp, P.G.; Corces, V.G. A subset of Drosophila Myc sites remain associated with mitotic chromosomes colocalized with insulator proteins. Nat. Commun. 2013, 4, 1464. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, H.D.; Jung, Y.; Kim, J.; Chung, J. Drosophila Low Temperature Viability Protein 1 (LTV1) Is Required for Ribosome Biogenesis and Cell Growth Downstream of Drosophila Myc (dMyc). J. Biol. Chem. 2015, 290, 13591–13604. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, N.C.; Tchoubrieva, E.B.; Chahal, A.; Woods, S.; Lee, A.; Lin, J.I.; Parsons, L.; Jastrzebski, K.; Poortinga, G.; Hannan, K.M.; et al. S6 Kinase is essential for MYC-dependent rDNA transcription in Drosophila. Cell. Signal. 2015, 27, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- de la Cova, C.; Abril, M.; Bellosta, P.; Gallant, P.; Johnston, L.A. Drosophila myc regulates organ size by inducing cell competition. Cell 2004, 117, 107–116. [Google Scholar] [CrossRef]
- Levens, D. Cellular MYCro economics: Balancing MYC function with MYC expression. Cold Spring Harbor Perspect. Med. 2013, 3, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Posternak, V.; Ung, M.H.; Cheng, C.; Cole, M.D. MYC Mediates mRNA Cap Methylation of Canonical Wnt/beta-catenin Signaling Transcripts by Recruiting CDK7 and RNA Methyltransferase. Mol. Cancer Res. 2016, 15, 213. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Sampson, V.B.; Rong, N.H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N.J.; Dunn, S.P.; Krueger, L.J. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007, 67, 9762–9770. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Saini, S.; Majid, S.; Hirata, H.; Ueno, K.; Deng, G.; Dahiya, R. MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS ONE 2012, 7, e29722. [Google Scholar] [CrossRef] [PubMed]
- Daneshvar, K.; Nath, S.; Khan, A.; Shover, W.; Richardson, C.; Goodliffe, J.M. MicroRNA miR-308 regulates dMyc through a negative feedback loop in Drosophila. Biol. Open 2013, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Zhao, X.; Tao, J. c-MYC-miRNA circuitry: A central regulator of aggressive B-cell malignancies. Cell Cycle 2014, 13, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Jackstadt, R.; Hermeking, H. MicroRNAs as regulators and mediators of c-MYC function. Biochim. Biophys. Acta 2015, 1849, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Herranz, H.; Hong, X.; Pérez, L.; Ferreira, A.; Olivieri, D.; Cohen, S.M.; Milán, M. The miRNA machinery targets Mei-P26 and regulates Myc protein levels in the Drosophila wing. EMBO J. 2010, 29, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Boulan, L.; Pérez, L.; Milán, M. Mei-P26 mediates tissue-specific responses to the Brat tumor suppressor and the dMyc proto-oncogene in Drosophila. Genetics 2014, 198, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 2009, 136, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Hann, S.R. c-Myc proteolysis by the ubiquitin-proteasome pathway: Stabilization of c-Myc in Burkitt’s lymphoma cells. Mol. Cell. Biol. 2000, 20, 2423–2435. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Lutterbach, B.; Hann, S.R. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol. Cell. Biol. 1994, 14, 5510–5522. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras enhances Myc protein stability. Mol. Cell 1999, 3, 169–179. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Grim, J.A.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [PubMed]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [PubMed]
- Parisi, F.; Riccardo, S.; Daniel, M.; Saqcena, M.; Kundu, N.; Pession, A.; Grifoni, D.; Stocker, H.; Tabak, E.; Bellosta, P. Drosophila insulin and target of rapamycin (TOR) pathways regulate GSK3 beta activity to control Myc stability and determine Myc expression in vivo. BMC Biol. 2011, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Pulverer, B.J.; Fisher, C.; Vousden, K.; Littlewood, T.; Evan, G.; Woodgett, J.R. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene 1994, 9, 59–70. [Google Scholar] [PubMed]
- Yano, T.; Sander, C.A.; Clark, H.M.; Dolezal, M.V.; Jaffe, E.S.; Raffeld, M. Clustered mutations in the second exon of the MYC gene in sporadic Burkitt’s lymphoma. Oncogene 1993, 8, 2741–2748. [Google Scholar] [PubMed]
- Flinn, E.M.; Busch, C.M.; Wright, A.P. myc boxes, which are conserved in myc family proteins, are signals for protein degradation via the proteasome. Mol. Cell. Biol. 1998, 18, 5961–5969. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, C.; Pan, J.; Wang, X.; Jin, J.; Zhao, L.; Pan, W.; Liao, G.; Cai, X.; Li, X.; et al. Regulation of c-Myc protein stability by proteasome activator REGγ. Cell Death Differ. 2015, 22, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Turner, S.A.; Cole, M.D. Burkitt's lymphoma-associated c-Myc mutations converge on a dramatically altered target gene response and implicate Nol5a/Nop56 in oncogenesis. Oncogene 2014, 33, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.; Cochran, B.H.; Stiles, C.D.; Leder, P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell 1983, 35, 603–610. [Google Scholar] [CrossRef]
- Grosso, L.E.; Pitot, H.C. Chromatin structure of the c-myc gene in HL-60 cells during alterations of transcriptional activity. Cancer Res. 1985, 45, 5035–5041. [Google Scholar] [PubMed]
- Bentley, D.L.; Groudine, M. A block to elongation is largely responsible for decreased transcription of c-myc in differentiated HL60 cells. Nature 1986, 321, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Duman-Scheel, M.; Johnston, L.A.; Du, W. Repression of dMyc expression by Wingless promotes Rbf-induced G1 arrest in the presumptive Drosophila wing margin. Proc. Natl. Acad. Sci. USA 2004, 101, 3857–3862. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Johnston, L.A. Control of wing size and proportions by Drosophila myc. Genetics 2010, 184, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Herranz, H.; Pérez, L.; Martín, F.A.; Milán, M. A Wingless and Notch double-repression mechanism regulates G1-S transition in the Drosophila wing. EMBO J. 2008, 27, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A.; Sanders, A.L. Wingless promotes cell survival but constrains growth during Drosophila wing development. Nat. Cell Biol. 2003, 5, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.J.; Wilkins, J.A.; Reed, K.R.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A.R. Myc deletion rescues APC deficiency in the small intestine. Nature 2007, 446, 676–679. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of APC in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Jay, P.; Gregorieff, A.; van Gijn, M.E.; Jonkheer, S.; Hatzis, P.; Thiele, A.; van den Born, M.; Begthel, H.; Brabletz, T.; et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat. Cell Biol. 2005, 7, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Andreu, P.; Colnot, S.; Godard, C.; Gad, S.; Chafey, P.; Niwa-Kawakita, M.; Laurent-Puig, P.; Kahn, A.; Robine, S.; Perret, C.; et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following APC loss in the mouse intestine. Development 2005, 132, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Pear, W.S.; Aster, J.C. The multifaceted role of Notch in cancer. Curr. Opin. Genet. Dev. 2007, 17, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Banerjee, S.; Sarkar, F.H. Exploitation of the Notch signaling pathway as a novel target for cancer therapy. Anticancer Res. 2008, 28, 3621–3630. [Google Scholar] [PubMed]
- De Celis, J.F.; Garcia-Bellido, A.; Bray, S.J. Activation and function of Notch at the dorsal-ventral boundary of the wing imaginal disc. Development 1996, 122, 359–369. [Google Scholar] [PubMed]
- Herranz, H.; Stamataki, E.; Feiguin, F.; Milán, M. Self-refinement of Notch activity through the transmembrane protein Crumbs: Modulation of gamma-secretase activity. EMBO Rep. 2006, 7, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Baonza, A.; Garcia-Bellido, A. Notch signaling directly controls cell proliferation in the Drosophila wing disc. Proc. Natl. Acad. Sci. USA 2000, 97, 2609–2614. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.J.; Cohen, S.M. A hierarchy of cross-regulation involving Notch, wingless, vestigial and cut organizes the dorsal/ventral axis of the Drosophila wing. Development 1996, 122, 3477–3485. [Google Scholar] [PubMed]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Djiane, A.; Krejci, A.; Bernard, F.; Fexova, S.; Millen, K.; Bray, S.J. Dissecting the mechanisms of Notch induced hyperplasia. EMBO J. 2013, 32, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Aradhya, R.; Zmojdzian, M.; Da Ponte, J.P.; Jagla, K. Muscle niche-driven Insulin-Notch-Myc cascade reactivates dormant Adult Muscle Precursors in Drosophila. Elife 2015, 4, e08497. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Liu, F.; Ye, B.; Zhang, X.; Liang, Y.; Yao, J. Notch4 promotes gastric cancer growth through activation of Wnt1/β-catenin signaling. Mol. Cell. Biochem. 2015, 401, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Klinakis, A.; Szabolcs, M.; Politi, K.; Kiaris, H.; Artavanis-Tsakonas, S.; Efstratiadis, A. Myc is a Notch1 transcriptional target and a requisite for Notch1-induced mammary tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9262–9267. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.M.; Calvo, J.A.; Draheim, K.M.; Cunningham, L.A.; Hermance, N.; Beverly, L.; Krishnamoorthy, V.; Bhasin, M.; Capobianco, A.J.; Kelliher, M.A. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol. Cell. Biol. 2006, 26, 8022–8031. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Einwächter, H.; Lee, M.; Sipos, B.; Nakhai, H.; Rad, R.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Klöppel, G.; et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2010, 107, 13438–13443. [Google Scholar] [CrossRef] [PubMed]
- Doumpas, N.; Ruiz-Romero, M.; Blanco, E.; Edgar, B.; Corominas, M.; Teleman, A.A. Brk regulates wing disc growth in part via repression of Myc expression. EMBO Rep. 2013, 14, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-R.; Kang, Y.; Siegel, P.M.; Massagué, J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 2002, 110, 19–32. [Google Scholar] [CrossRef]
- Ren, F.; Shi, Q.; Chen, Y.; Jiang, A.; Ip, Y.T.; Jiang, H.; Jiang, J. Drosophila Myc integrates multiple signaling pathways to regulate intestinal stem cell proliferation during midgut regeneration. Cell Res. 2013, 23, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Drennan, A.C.; Ceribelli, M.; Zhu, F.; Wright, G.W.; Huang, D.W.; Xiao, W.; Li, Y.; Grindle, K.M.; Lu, L.; et al. Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E7260–E7267. [Google Scholar] [CrossRef] [PubMed]
- Teleman, A.A.; Hietakangas, V.; Sayadian, A.C.; Cohen, S.M. Nutritional control of protein biosynthetic capacity by insulin via Myc in Drosophila. Cell Metable 2008, 7, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Neto-Silva, R.M.; de Beco, S.; Johnston, L.A. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev. Cell 2010, 19, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, J.; Ou, C.; Zhang, Y.; Ma, L.; Weng, W.; Pan, Q.; Sun, F. Mutual interaction between YAP and c-Myc is critical for carcinogenesis in liver cancer. Biochem. Biophys. Res. Commun. 2013, 439, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, P.G.; Knowles, D.M.; Magrath, I.; Dalla-Favera, R. Chromosomal breakpoints and structural alterations of the c-myc locus differ in endemic and sporadic forms of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1986, 83, 2984–2988. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; LeStrange, R.C.; Novak, U.; Hayward, W.S.; Groudine, M. The block to transcription elongation is promoter dependent in normal and Burkitt's lymphoma c-myc alleles. Genes Dev. 1990, 4, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; Groudine, M. Molecular analysis of the c-myc transcription elongation block. Implications for the generation of Burkitt’s lymphoma. Ann. N. Y. Acad. Sci. 1990, 599, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; Groudine, M. Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res. 1991, 56, 1–48. [Google Scholar] [PubMed]
- Siebenlist, U.; Hennighausen, L.; Battey, J.; Leder, P. Chromatin structure and protein binding in the putative regulatory region of the c-myc gene in Burkitt lymphoma. Cell 1984, 37, 381–391. [Google Scholar] [CrossRef]
- Schneider, E.E.; Albert, T.; Wolf, D.A.; Eick, D. Regulation of c-myc and immunoglobulin kappa gene transcription by promoter-proximal pausing of RNA polymerase II. Curr. Top. Microbiol. Immunol. 1999, 246, 225–231. [Google Scholar] [PubMed]
- Krumm, A.; Hickey, L.B.; Groudine, M. Promoter-proximal pausing of RNA polymerase II defines a general rate-limiting step after transcription initiation. Genes Dev. 1995, 9, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Grosso, L.E.; Pitot, H.C. Transcriptional regulation of c-myc during chemically induced differentiation of HL-60 cultures. Cancer Res. 1985, 45, 847–850. [Google Scholar] [PubMed]
- Avigan, M.I.; Strober, B.; Levens, D. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J. Biol. Chem. 1990, 265, 18538–18545. [Google Scholar] [PubMed]
- Duncan, R.; Bazar, L.; Michelotti, G.; Tomonaga, T.; Krutzsch, H.; Avigan, M.; Levens, D. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994, 8, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Michelotti, E.F.; Pullner, A.; Duncan, R.C.; Eick, D.; Levens, D. Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol. Cell. Biol. 1996, 16, 2656–2669. [Google Scholar] [CrossRef] [PubMed]
- Krumm, A.; Meulia, T.; Brunvand, M.; Groudine, M. The block to transcriptional elongation within the human c-myc gene is determined in the promoter-proximal region. Genes Dev. 1992, 6, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Bazar, L.; Meighen, D.; Harris, V.; Duncan, R.; Levens, D.; Avigan, M. Targeted melting and binding of a DNA regulatory element by a transactivator of c-myc. J. Biol. Chem. 1995, 270, 8241–8248. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Sanford, S.; Elisha-Feil, Z.; Levens, D. The functional response of upstream DNA to dynamic supercoiling in vivo. Nat. Struct. Mol. Biol. 2008, 15, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Levens, D. How the c-myc promoter works and why it sometimes does not. J. Natl. Cancer Inst. Monogr. 2008, 2008, 41–43. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liu, J.; Collins, I.; Sanford, S.; O'Connell, B.; Benham, C.J.; Levens, D. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 2000, 19, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Rabenhorst, U.; Beinoraviciute-Kellner, R.; Brezniceanu, M.-L.; Joos, S.; Devens, F.; Lichter, P.; Rieker, R.J.; Trojan, J.; Chung, H.-J.; Levens, D.L.; et al. Overexpression of the far upstream element binding protein 1 in hepatocellular carcinoma is required for tumor growth. Hepatology 2009, 50, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Rabenhorst, U.; Thalheimer, F.B.; Gerlach, K.; Kijonka, M.; Böhm, S.; Krause, D.S.; Vauti, F.; Arnold, H.-H.; Schroeder, T.; Schnütgen, F.; et al. Single-Stranded DNA-Binding Transcriptional Regulator FUBP1 Is Essential for Fetal and Adult Hematopoietic Stem Cell Self-Renewal. Cell Rep. 2015, 11, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A.L. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chung, Y.-J.; Castellar, E.R.P.; Zheng, Y.; Chung, H.-J.; Bandle, R.; Liu, J.; Tessarollo, L.; Batchelor, E.; Aplan, P.D.; et al. Far Upstream Element Binding Protein Plays aCrucial Role in Embryonic Development, Hematopoiesis, and Stabilizing MYC Expression Levels. Am. J. Pathol. 2016, 186, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Davis-Smyth, T.; Duncan, R.C.; Zheng, T.; Michelotti, G.; Levens, D. The far upstream element-binding proteins comprise an ancient family of single-strand DNA-binding transactivators. J. Biol. Chem. 1996, 271, 31679–31687. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.-J.; Liu, J.; Dundr, M.; Nie, Z.; Sanford, S.; Levens, D. FBPs are calibrated molecular tools to adjust gene expression. Mol. Cell. Biol. 2006, 26, 6584–6597. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, L.; Collins, I.; Ge, H.; Libutti, D.; Li, J.; Egly, J.M.; Levens, D. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol. Cell 2000, 5, 331–341. [Google Scholar] [CrossRef]
- Hahn, S. Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol. 2004, 11, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Fazal, F.M.; Meng, C.A.; Murakami, K.; Kornberg, R.D.; Block, S.M. Real-time observation of the initiation of RNA polymerase II transcription. Nature 2015, 525, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Buratowski, S. Structural biology: Snapshots of transcription initiation. Nature 2016, 533, 331. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Ohkuma, Y. Phosphorylation of the C-terminal domain of RNA polymerase II plays central roles in the integrated events of eucaryotic gene expression. J. Biochem. 2007, 141, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Larivière, L.; Seizl, M.; Cramer, P. A structural perspective on Mediator function. Curr. Opin. Cell Biol. 2012, 24, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.H.; Jin, Y.; Struhl, K. TFIIH Phosphorylation of the Pol II CTD Stimulates Mediator Dissociation from the Preinitiation Complex and Promoter Escape. Mol. Cell 2014, 54, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Drapkin, R.; Reinberg, D. The multifunctional TFIIH complex and transcriptional control. Trends Biochem. Sci. 1994, 19, 504–508. [Google Scholar] [CrossRef]
- Tirode, F.; Busso, D.; Coin, F.; Egly, J.M. Reconstitution of the transcription factor TFIIH: Assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol. Cell 1999, 3, 87–95. [Google Scholar] [CrossRef]
- Bradsher, J.; Coin, F.; Egly, J.M. Distinct roles for the helicases of TFIIH in transcript initiation and promoter escape. J. Biol. Chem. 2000, 275, 2532–2538. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kouzine, F.; Nie, Z.; Chung, H.-J.; Elisha-Feil, Z.; Weber, A.; Zhao, K.; Levens, D. The FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. EMBO J. 2006, 25, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Tomonaga, T.; Shimada, H.; Shioya, A.; Higashi, M.; Matsubara, H.; Harigaya, K.; Nomura, F.; Libutti, D.; Levens, D.; et al. An essential role of alternative splicing of c-myc suppressor FUSE-binding protein-interacting repressor in carcinogenesis. Cancer Res. 2006, 66, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Compe, E.; Egly, J.-M. TFIIH: When transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 2012, 13, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Coin, F.; Bergmann, E.; Tremeau-Bravard, A.; Egly, J.M. Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J. 1999, 18, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Akoulitchev, S.; Weber, A.; Ge, H.; Chuikov, S.; Libutti, D.; Wang, X.W.; Conaway, J.W.; Harris, C.C.; Conaway, R.C.; et al. Defective interplay of activators and repressors with TFIH in xeroderma pigmentosum. Cell 2001, 104, 353–363. [Google Scholar] [CrossRef]
- Oh, K.-S.; Khan, S.G.; Jaspers, N.G.J.; Raams, A.; Ueda, T.; Lehmann, A.; Friedmann, P.S.; Emmert, S.; Gratchev, A.; Lachlan, K.; et al. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum. Mutat. 2006, 27, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.M.; Dickins, R.A.; Coombe, M.; Hime, G.R.; Bowtell, D.D.L.; Richardson, H. Drosophila Hfp negatively regulates dmyc and stg to inhibit cell proliferation. Development 2004, 131, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, N.C.; Johanson, T.M.; Cranna, N.J.; Er, A.L.J.; Richardson, H.E.; Hannan, R.D.; Quinn, L.M. Hfp inhibits Drosophila myc transcription and cell growth in a TFIIH/Hay-dependent manner. Development 2010, 137, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.C.; Jones, R.S.; Liang, B.C.; Gelbart, W.; Fuller, M.T. A Drosophila model for xeroderma pigmentosum and Cockayne’s syndrome: Haywire encodes the fly homolog of ERCC3, a human excision repair gene. Cell 1992, 71, 925–937. [Google Scholar] [CrossRef]
- Mounkes, L.C.; Fuller, M.T. Molecular characterization of mutant alleles of the DNA repair/basal transcription factor haywire/ERCC3 in Drosophila. Genetics 1999, 152, 291–297. [Google Scholar] [PubMed]
- Merino, C.; Reynaud, E.; Vázquez, M.; Zurita, M. DNA repair and transcriptional effects of mutations in TFIIH in Drosophila development. Mol. Biol. Cell 2002, 13, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.A.; Mitchell, N.C.; Zaytseva, O.; Chahal, A.; Mendis, P.; Cartier-Michaud, A.; Parsons, L.M.; Poortinga, G.; Levens, D.L.; Hannan, R.D.; et al. Defective Hfp-dependent transcriptional repression of dMYC is fundamental to tissue overgrowth in Drosophila XPB models. Nat. Commun. 2015, 6, 7404. [Google Scholar] [CrossRef] [PubMed]
- Gallant, P. Drosophila Myc. Adv. Cancer Res. 2009, 103, 111–144. [Google Scholar] [PubMed]
- Siebel, C.W.; Kanaar, R.; Rio, D.C. Regulation of tissue-specific P-element pre-mRNA splicing requires the RNA-binding protein PSI. Genes Dev. 1994, 8, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Brookfield, J.F.; Montgomery, E.; Langley, C.H. Apparent absence of transposable elements related to the P elements of D. melanogaster in other species of Drosophila. Nature 1984, 310, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Taliaferro, J.M.; Klibaite, U.; Hilgers, V.; Shaevitz, J.W.; Rio, D.C. The PSI-U1 snRNP interaction regulates male mating behavior in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, 5269–5274. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zaysteva, O.; Nie, Z.; Mitchell, N.C.; Amanda Lee, J.E.; Ware, T.; Parsons, L.; Luwor, R.; Poortinga, G.; Hannan, R.D.; et al. Defining the essential function of FBP/KSRP proteins: Drosophila Psi interacts with the mediator complex to modulate MYC transcription and tissue growth. Nucleic Acids Res. 2016, 44, 7646–7658. [Google Scholar] [CrossRef] [PubMed]
- Guruharsha, K.G.; Rual, J.-F.; Zhai, B.; Mintseris, J.; Vaidya, P.; Vaidya, N.; Beekman, C.; Wong, C.; Rhee, D.Y.; Cenaj, O.; et al. A protein complex network of Drosophila melanogaster. Cell 2011, 147, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Collins, I.; Tomonaga, T.; Zhang, T.; Levens, D. A unique transactivation sequence motif is found in the carboxyl-terminal domain of the single-strand-binding protein FBP. Mol. Cell. Biol. 1996, 16, 2274–2282. [Google Scholar] [CrossRef] [PubMed]
- Boube, M.; Joulia, L.; Cribbs, D.L.; Bourbon, H.-M. Evidence for a mediator of RNA polymerase II transcriptional regulation conserved from yeast to man. Cell 2002, 110, 143–151. [Google Scholar] [CrossRef]
- Bourbon, H.-M. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 2008, 36, 3993–4008. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.W.; Veschambre, P.; Erdjument-Bromage, H.; Tempst, P.; Conaway, J.W.; Conaway, R.C.; Kornberg, R.D. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 8538–8543. [Google Scholar] [CrossRef] [PubMed]
- Taatjes, D.J. The human Mediator complex: A versatile, genome-wide regulator of transcription. Trends Biochem. Sci. 2010, 35, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A centralintegrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Van de Peppel, J.; Kettelarij, N.; van Bakel, H.; Kockelkorn, T.T.J.P.; van Leenen, D.; Holstege, F.C.P. Mediator expression profiling epistasis reveals a signal transduction pathway with antagonistic submodules and highly specific downstream targets. Mol. Cell 2005, 19, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Xu, X.; Hecht, A.; Boyer, T.G. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem. 2006, 281, 14066–14075. [Google Scholar] [CrossRef] [PubMed]
- Carrera, I.; Janody, F.; Leeds, N.; Duveau, F.; Treisman, J.E. Pygopus activates Wingless target gene transcription through the mediator complex subunits Med12 and Med13. Proc. Natl. Acad. Sci. USA 2008, 105, 6644–6649. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Janody, F.; Martirosyan, Z.; Benlali, A.; Treisman, J.E. Two subunits of the Drosophila mediator complex act together to control cell affinity. Development 2003, 130, 3691–3701. [Google Scholar] [CrossRef] [PubMed]
- Janody, F.; Treisman, J.E. Requirements for mediator complex subunits distinguish three classes of notch target genes at the Drosophila wing margin. Dev. Dyn. 2011, 240, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Marr, S.K.; Lis, J.T.; Treisman, J.E.; Marr, M.T. The metazoan-specific mediator subunit 26 (Med26) is essential for viability and is found at both active genes and pericentric heterochromatin in Drosophila melanogaster. Mol. Cell. Biol. 2014, 34, 2710–2720. [Google Scholar] [CrossRef] [PubMed]
- Boube, M.; Hudry, B.; Immarigeon, C.; Carrier, Y.; Bernat-Fabre, S.; Merabet, S.; Graba, Y.; Bourbon, H.-M.; Cribbs, D.L. Drosophila melanogaster Hox transcription factors access the RNA polymerase II machinery through direct homeodomain binding to a conserved motif of mediator subunit Med19. PLoS Genet. 2014, 10, e1004303. [Google Scholar] [CrossRef] [PubMed]
- Terriente-Félix, A.; López-Varea, A.; de Celis, J.F. Identification of genes affecting wing patterning through a loss-of-function mutagenesis screen and characterization of med15 function during wing development. Genetics 2010, 185, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Yang, X.; Fu, L.; Lv, X.; Zhang, Z.; Wu, W.; Yang, S.; Zhou, Z.; Zhang, L.; Zhao, Y. The Kto-Skd complex can regulate ptc expression by interacting with Cubitus interruptus (Ci) in the Hedgehog signaling pathway. J. Biol. Chem. 2014, 289, 22333–22341. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Li, L.; Qin, Z.S.; Corces, V.G. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell 2012, 48, 471–484. [Google Scholar] [CrossRef] [PubMed]
- De Laat, W.; Dekker, J. 3C-based technologies to study the shape of the genome. Methods 2012, 58, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.-T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Day, D.S.; Young, R.A. Insulated Neighborhoods: Structuraland Functional Units of Mammalian Gene Control. Cell 2016, 167, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Chromosomes: Dynamically in the loop. Nat. Rev. Genet. 2014, 15, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Fay, A.; Misulovin, Z.; Li, J.; Schaaf, C.A.; Gause, M.; Gilmour, D.S.; Dorsett, D. Cohesin selectively binds and regulates genes with paused RNA polymerase. Curr. Biol. 2011, 21, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, E.F.; Sanford, S.; Levens, D. Marking of active genes on mitotic chromosomes. Nature 1997, 388, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Wang, D.; Ye, B.; Shi, M.; Zhang, Y.; Zhao, Z. A possible role of Drosophila CTCF in mitotic bookmarking and maintaining chromatin domains during the cell cycle. Biol. Res. 2015, 48, 27. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, T.; Lim, B.; Levine, M. Enhancer Control of Transcriptional Bursting. Cell 2016, 166, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Lobanenkov, V.V.; Nicolas, R.H.; Adler, V.V.; Paterson, H.; Klenova, E.M.; Polotskaja, A.V.; Goodwin, G.H. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 1990, 5, 1743–1753. [Google Scholar] [PubMed]
- Rhodes, J.M.; Bentley, F.K.; Print, C.G.; Dorsett, D.; Misulovin, Z.; Dickinson, E.J.; Crosier, K.E.; Crosier, P.S.; Horsfield, J.A. Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev. Biol. 2010, 344, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Esposito, D.; Duhagon, M.A.; Banfield, K.; Mehalko, J.; Liao, H.; Stephens, R.M.; Harris, T.J.R.; Munroe, D.J.; Wu, X. Long-range enhancers on 8q24 regulate c-Myc. Proc. Natl. Acad. Sci. USA 2010, 107, 3001–3005. [Google Scholar] [CrossRef] [PubMed]
- Uslu, V.V.; Petretich, M.; Ruf, S.; Langenfeld, K.; Fonseca, N.A.; Marioni, J.C.; Spitz, F. Long-range enhancers regulating Myc expression are required for normal facial morphogenesis. Nat. Genet. 2014, 46, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. Elife 2016, 5, e18270. [Google Scholar] [CrossRef] [PubMed]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.S.; et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Shaw, A.T. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J. Clin. Oncol. 2013, 31, 3987–3996. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.; Michor, F. Evolution of acquired resistance to anti-cancer therapy. J. Theor. Biol. 2014, 355, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Jucker, R.; Panacchia, L.; Ricordy, R.; Tatò, F.; Nasi, S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 2002, 62, 3507–3510. [Google Scholar] [PubMed]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Annibali, D.; Whitfield, J.R.; Favuzzi, E.; Jauset, T.; Serrano, E.; Cuartas, I.; Redondo-Campos, S.; Folch, G.; Gonzàlez-Juncà, A.; Sodir, N.M.; et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 2014, 5, 4632. [Google Scholar] [CrossRef] [PubMed]
- Posternak, V.; Cole, M.D. Strategically targeting MYC in cancer. F1000Res 2016, 5, 408. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M. The Quest for Targets Executing MYC-Dependent Cell Transformation. Front Oncol 2016, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.-S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition ofTumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Knoechel, B.; Roderick, J.E.; Williamson, K.E.; Zhu, J.; Lohr, J.G.; Cotton, M.J.; Gillespie, S.M.; Fernandez, D.; Ku, M.; Wang, H.; et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J.; Singer, D.S. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.C.; Debrosse, M.; Smith, M.; Dey, A.; Huynh, W.; Sarai, N.; Heightman, T.D.; Tamura, T.; Ozato, K. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol. Cell. Biol. 2013, 33, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, A.S.; Roe, J.-S.; Mok, B.Y.L.; Hohmann, A.F.; Shi, J.; Vakoc, C.R. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep. 2016, 15, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Fernando, C.; Audibert, A.; Simon, F.; Tazi, J.; Juge, F. A role for the serine/arginine-rich (SR) protein B52/SRSF6 in cell growth and myc expression in Drosophila. Genetics 2015, 199, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Ring, H.Z.; Lis, J.T. The SR protein B52/SRp55 is essential for Drosophila development. Mol. Cell. Biol. 1994, 14, 7499–7506. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaytseva, O.; Quinn, L.M. Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling. Genes 2017, 8, 118. https://doi.org/10.3390/genes8040118
Zaytseva O, Quinn LM. Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling. Genes. 2017; 8(4):118. https://doi.org/10.3390/genes8040118
Chicago/Turabian StyleZaytseva, Olga, and Leonie M. Quinn. 2017. "Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling" Genes 8, no. 4: 118. https://doi.org/10.3390/genes8040118
APA StyleZaytseva, O., & Quinn, L. M. (2017). Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling. Genes, 8(4), 118. https://doi.org/10.3390/genes8040118