
Splice-Switching Therapy for Spinal Muscular Atrophy

Abstract
:1. Introduction
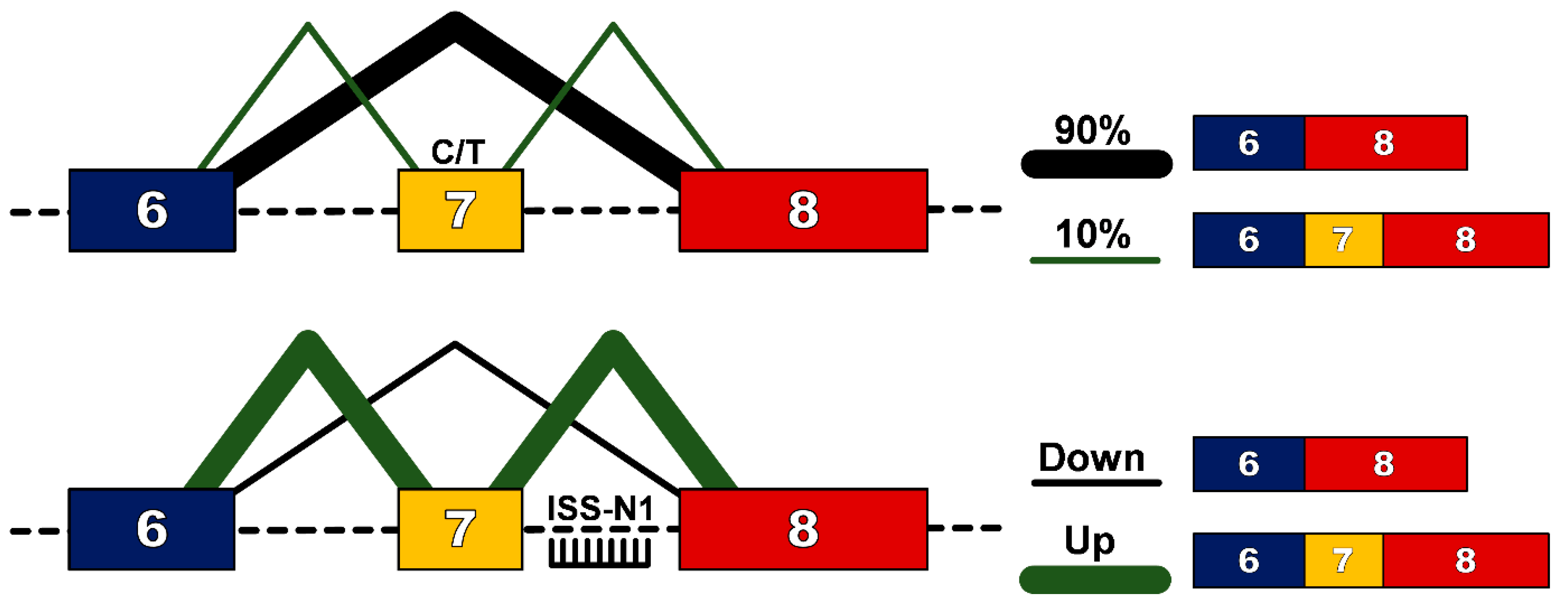
2. Regulation of Alternative Splicing in Exon 7 of SMN2 Gene
3. Clinical Trials of SSOs in SMA
4. Challenges for SSOs for the Treatment of SMA
5. Non-SSO Strategies SMN Up-Regulation
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ben-Shachar, S.; Orr-Urtreger, A.; Bardugo, E.; Shomrat, R.; Yaron, Y. Large-scale population screening for spinal muscular atrophy: Clinical implications. Genet. Med. 2011, 13, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Miniño, A.M.; Xu, J.; Kochanek, K.D. National Vital Statistics Reports; U.S. Department of Health and Human Services: Washington, DC, USA, 2008; Volume 59, Number 2.
- Wadman, R.I.; Vrancken, A.F.J.E.; van den Berg, L.H.; van der Pol, W.L. Dysfunction of the neuromuscular junction in spinal muscular atrophy types 2 and 3. Neurology 2012, 79, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Pardo, C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Pellizzoni, L.; Yong, J.; Dreyfuss, G. Essential role for the SMN complex in the specificity of snRNP assembly. Science 2002, 298, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Workman, E.; Kolb, S.J.; Battle, D.J. Spliceosomal small nuclear ribonucleoprotein biogenesis defects and motor neuron selectivity in spinal muscular atrophy. Brain Res. 2012, 1462, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Gavrilina, T.O.; McGovern, V.L.; Workman, E.; Crawford, T.O.; Gogliotti, R.G.; DiDonato, C.J.; Monani, U.R.; Morris, G.E.; Burghes, A.H.M. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 2008, 17, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Oprea, G.E.; Kröber, S.; McWhorter, M.L.; Rossoll, W.; Müller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Yener, İ.H.; Topaloglu, H.; Erdem-Özdamar, S.; Dayangac-Erden, D. Transcript levels of plastin 3 and neuritin 1 modifier genes in spinal muscular atrophy siblings. Pediatr. Int. 2017, 59, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Androphy, E.J.; Singh, R.N. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA 2004, 10, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.-D.; Ares, M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Kashima, T.; Manley, J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Pedrotti, S.; Bielli, P.; Paronetto, M.P.; Ciccosanti, F.; Fimia, G.M.; Stamm, S.; Manley, J.L.; Sette, C. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 2010, 29, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Androphy, E.J.; Singh, R.N. An extended inhibitory context causes skipping of exon 7 of SMN2 in spinal muscular atrophy. Biochem. Biophys. Res. Commun. 2004, 315, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.R.; Hertel, K.J. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J. Biol. Chem. 2001, 276, 45476–45483. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.K.; Singh, N.N.K.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.H.; Schray, R.C.; Patterson, C.A.; Ayitey, S.O.; Tallent, M.K.; Lutz, G.J. Oligonucleotide-Mediated Survival of Motor Neuron Protein Expression in CNS Improves Phenotype in a Mouse Model of Spinal Muscular Atrophy. J. Neurosci. 2009, 29, 7633–7638. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [PubMed]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Janghra, N.; Mitrpant, C.; Dickinson, R.L.; Anthony, K.; Price, L.; Eperon, I.C.; Wilton, S.D.; Morgan, J.; Muntoni, F. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum. Gene Ther. 2013, 24, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; McGovern, V.L.; Alwine, I.E.; Wang, X.; Massoni-Laporte, A.; Rich, M.M.; Burghes, A.H.M. Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 2011, 20, 3578–3591. [Google Scholar] [CrossRef] [PubMed]
- Robbins, K.L.; Glascock, J.J.; Osman, E.Y.; Miller, M.R.; Lorson, C.L. Defining the therapeutic window in a severe animal model of spinal muscular atrophy. Hum. Mol. Genet. 2014, 23, 4559–4568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Meng, J.; Marrosu, E.; Janghra, N.; Morgan, J.; Muntoni, F. Repeated low doses of morpholino antisense oligomer: An intermediate mouse model of spinal muscular atrophy to explore the window of therapeutic response. Hum. Mol. Genet. 2015, 24, 6265–6277. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Baker, B.F.; Bennett, C.F.; Krainer, A.R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007, 5, e73. [Google Scholar] [CrossRef] [PubMed]
- Skordis, L.A.; Dunckley, M.G.; Yue, B.; Eperon, I.C.; Muntoni, F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl. Acad. Sci. USA 2003, 100, 4114–4119. [Google Scholar] [CrossRef] [PubMed]
- Osman, E.Y.; Yen, P.-F.; Lorson, C.L. Bifunctional RNAs Targeting the Intronic Splicing Silencer N1 Increase SMN Levels and Reduce Disease Severity in an Animal Model of Spinal Muscular Atrophy. Mol. Ther. 2012, 20, 119–126. [Google Scholar] [CrossRef] [PubMed]
- d’Ydewalle, C.; Ramos, D.M.; Pyles, N.J.; Ng, S.-Y.; Gorz, M.; Pilato, C.M.; Ling, K.; Kong, L.; Ward, A.J.; Rubin, L.L.; et al. The Antisense Transcript SMN-AS1 Regulates SMN Expression and Is a Novel Therapeutic Target for Spinal Muscular Atrophy. Neuron 2017, 93, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Darras, B.; Chiriboga, C.; Swoboda, K.; Iannaccone, S.; Montes, J.; Castro, D.; Holuba, N.; Raush, N.; Visyak, N.; Dunaway, S.; et al. G.O.18: Results of a phase 2 study of ISIS-SMNRx in children with spinal muscular atrophy. Neuromuscul. Disord. 2014, 24, 920. [Google Scholar] [CrossRef]
- Biogen and Ionis Pharmaceuticals Announce SPINRAZA (nusinersen) Meets Primary Endpoint at Interim Analysis of Phase 3 CHERISH Study in Later-Onset Spinal Muscular Atrophy. Available online: http://ir.ionispharma.com/phoenix.zhtml?c=222170&p=irol-newsArticle&ID=2220037 (accessed 10 March 2017).
- U.S. FDA Approves Biogen’s SPINRAZA™ (nusinersen), The First Treatment for Spinal Muscular Atrophy, Biogen Media. Available online: http://media.biogen.com/press-release/neurodegenerative-diseases/us-fda-approves-biogens-spinraza-nusinersen-first-treatment (accessed on 24 December 2016).
- Haché, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J. Child Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schoneborn, S.; Heller, R.; Berg, C.; Betzler, C.; Grimm, T.; Eggermann, T.; Eggermann, K.; Wirth, R.; Wirth, B.; Zerres, K. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet. 2008, 45, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Shababi, M.; Lorson, C.L.; Rudnik-Schöneborn, S.S. Spinal muscular atrophy: A motor neuron disorder or a multi-organ disease? J. Anat. 2014, 224, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Nizzardo, M.; Simone, C.; Salani, S.; Ruepp, M.-D.; Rizzo, F.; Ruggieri, M.; Zanetta, C.; Brajkovic, S.; Moulton, H.M.; Müehlemann, O.; et al. Effect of Combined Systemic and Local Morpholino Treatment on the Spinal Muscular Atrophy Δ7 Mouse Model Phenotype. Clin. Ther. 2014, 36, 340–356.e5. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.; Devi, G.R.; Iversen, P.L. Neutrally charged phosphorodiamidate morpholino antisense oligomers: uptake, efficacy and pharmacokinetics. Curr. Pharm. Biotechnol. 2004, 5, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates With Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol. Ther. Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Kayali, R.; Bertoni, C.; Fike, F.; Hu, H.; Iversen, P.L.; Gatti, R.A. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011, 20, 3151–3160. [Google Scholar] [CrossRef] [PubMed]
- Shabanpoor, F.; Hammond, S.M.; Abendroth, F.; Hazell, G.; Wood, M.J.A.; Gait, M.J. Identification of a Peptide for Systemic Brain Delivery of a Morpholino Oligonucleotide in Mouse Models of Spinal Muscular Atrophy. Nucl. Acid Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug. Chem. 2007, 18, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Deas, T.S.; Bennett, C.J.; Jones, S.A.; Tilgner, M.; Ren, P.; Behr, M.J.; Stein, D.A.; Iversen, P.L.; Kramer, L.D.; Bernard, K.A.; et al. In vitro resistance selection and in vivo efficacy of morpholino oligomers against West Nile virus. Antimicrob. Agents Chemother. 2007, 51, 2470–2482. [Google Scholar] [CrossRef] [PubMed]
- Glascock, J.J.; Shababi, M.; Wetz, M.J.; Krogman, M.M.; Lorson, C.L. Direct central nervous system delivery provides enhanced protection following vector mediated gene replacement in a severe model of spinal muscular atrophy. Biochem. Biophys. Res. Commun. 2012, 417, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Roskelley, E.M.; Richards, A.M.; Sardi, S.P.; O‘Riordan, C.R.; Klinger, K.W.; Shihabuddin, L.S.; Cheng, S.H. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Investig. 2010, 120, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Duque, S.I.; Arnold, W.D.; Odermatt, P.; Li, X.; Porensky, P.N.; Schmelzer, L.; Meyer, K.; Kolb, S.J.; Schümperli, D.; Kaspar, B.K.; et al. A large animal model of spinal muscular atrophy and correction of phenotype. Ann. Neurol. 2015, 77, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Glanzman, A.M.; Mazzone, E.; Main, M.; Pelliccioni, M.; Wood, J.; Swoboda, K.J.; Scott, C.; Pane, M.; Messina, S.; Bertini, E.; et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test development and reliability. Neuromuscul. Disord. 2010, 20, 155–161. [Google Scholar] [CrossRef] [PubMed]
- AveXis Reports Topline Results from Phase 1 Trial of AVXS-101 in SMA Type 1 and Fourth Quarter and Full Year 2016 Financial and Operating Results. Available online: http://investors.avexis.com/phoenix.zhtml?c=254285&p=irol-newsArticle&ID=2254564 (accessed on 20 April 2017).
- Basner-Tschakarjan, E.; Mingozzi, F. Cell-Mediated Immunity to AAV Vectors, Evolving Concepts and Potential Solutions. Front. Immunol. 2014, 5, 350. [Google Scholar] [CrossRef] [PubMed]
- Sumner, C.J.; Huynh, T.N.; Markowitz, J.A.; Perhac, J.S.; Hill, B.; Coovert, D.D.; Schussler, K.; Chen, X.; Jarecki, J.; Burghes, A.H.M.; et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol. 2003, 54, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, K.J.; Scott, C.B.; Reyna, S.P.; Prior, T.W.; LaSalle, B.; Sorenson, S.L.; Wood, J.; Acsadi, G.; Crawford, T.O.; Kissel, J.T.; et al. Phase II Open Label Study of Valproic Acid in Spinal Muscular Atrophy. PLoS ONE 2009, 4, e5268. [Google Scholar] [CrossRef] [PubMed]
- Weihl, C.C.; Connolly, A.M.; Pestronk, A. Valproate may improve strength and function in patients with type III/IV spinal muscle atrophy. Neurology 2006, 67, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Kissel, J.T.; Elsheikh, B.; King, W.M.; Freimer, M.; Scott, C.B.; Kolb, S.J.; Reyna, S.P.; Crawford, T.O.; Simard, L.R.; Krosschell, K.J.; et al. SMA valiant trial: A prospective, double-blind, placebo-controlled trial of valproic acid in ambulatory adults with spinal muscular atrophy. Muscle Nerve 2014, 49, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Kissel, J.T.; Scott, C.B.; Reyna, S.P.; Crawford, T.O.; Simard, L.R.; Krosschell, K.J.; Acsadi, G.; Elsheik, B.; Schroth, M.K.; D‘Anjou, G.; et al. SMA CARNIVAL TRIAL PART II: A prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS ONE 2011, 6, e21296. [Google Scholar] [CrossRef] [PubMed]
- Pruss, R.M.; Giraudon-Paoli, M.; Morozova, S.; Berna, P.; Abitbol, J.-L.; Bordet, T. Drug discovery and development for spinal muscular atrophy: Lessons from screening approaches and future challenges for clinical development. Future Med. Chem. 2010, 2, 1429–1440. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type | Age of Onset | Symptoms | Life Expectancy | SMN2 Copies |
|---|---|---|---|---|
| 0 | Prenatal | Less active foetus | <birth | 1 |
| 1 | 0–6 months | Cannot sit, respiratory muscle weakness | <2 years | 2 |
| 2 | 6–18 months | Cannot walk | <40 years | 3, 4 |
| 3 | 18 months–5 years | Needs support to walk | Adult | 3, 4 |
| 4 | >5 years | Restricted mobility | Adult | 4–8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meijboom, K.E.; Wood, M.J.A.; McClorey, G. Splice-Switching Therapy for Spinal Muscular Atrophy. Genes 2017, 8, 161. https://doi.org/10.3390/genes8060161
Meijboom KE, Wood MJA, McClorey G. Splice-Switching Therapy for Spinal Muscular Atrophy. Genes. 2017; 8(6):161. https://doi.org/10.3390/genes8060161
Chicago/Turabian StyleMeijboom, Katharina E., Matthew J.A. Wood, and Graham McClorey. 2017. "Splice-Switching Therapy for Spinal Muscular Atrophy" Genes 8, no. 6: 161. https://doi.org/10.3390/genes8060161
APA StyleMeijboom, K. E., Wood, M. J. A., & McClorey, G. (2017). Splice-Switching Therapy for Spinal Muscular Atrophy. Genes, 8(6), 161. https://doi.org/10.3390/genes8060161