
The Role of Replication-Associated Repair Factors on R-Loops



{kind=link}
Abstract
:1. Introduction
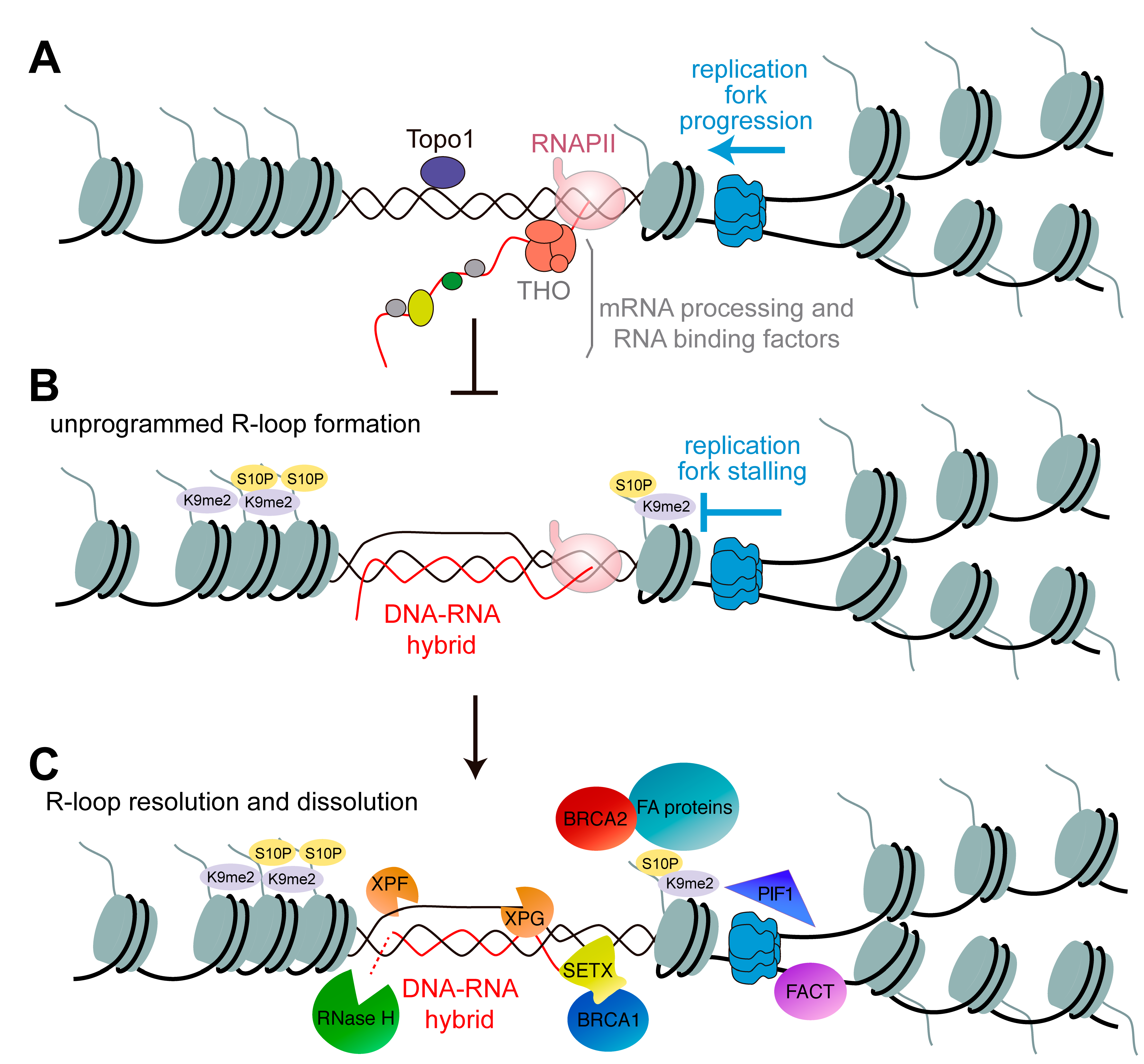
2. Preventing R-Loop Formation and Accumulation
3. Impact of R-Loops on Chromatin Structure
4. DNA Replication and Repair in the Context of R-Loops
5. Role of BRCA Genes in R-Loop Resolution
6. Role of the Fanconi Anemia Repair Pathway in R-Loop Resolution
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Rich, A. A hybrid helix containing both deoxyribose and ribose polynucleotides and its relation to the transfer of information between the nucleic acids. Proc. Natl. Acad. Sci. USA 1960, 46, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Gall, J.G.; Pardue, M.L. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc. Natl. Acad. Sci. USA 1969, 63, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to double-stranded DNA: Formation of R-loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote XRN2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Costantino, L.; Tan, F.J.; Zimmer, A.; Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 2016, 30, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chedin, F. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Houlard, M.; Artus, J.; Leguillier, T.; Vandormael-Pournin, S.; Cohen-Tannoudji, M. DNA-RNA hybrids contribute to the replication dependent genomic instability induced by Omcg1 deficiency. Cell Cycle 2011, 10, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Merrikh, H.; Zhang, Y.; Grossman, A.D.; Wang, J.D. Replication-transcription conflicts in bacteria. Nat. Rev. Microbiol. 2012, 10, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, S.; Crabbe, L.; Conti, C.; Tourriere, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Wellinger, R.E.; Prado, F.; Aguilera, A. Replication fork progression is impaired by transcription in hyperrecombinant yeast cells lacking a functional THO complex. Mol. Cell Biol. 2006, 26, 3327–3334. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Garcia-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. Conflict resolution in the genome: How transcription and replication make it work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; Garcia-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 facilitates replication through common fragile sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Sollier, J.; Cimprich, K.A. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015, 25, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Pozo, M.; Garcia-Muse, T.; Aguilera, A. R-loops cause replication impairment and genome instability during meiosis. EMBO Rep. 2012, 13, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Manley, J.L. Inactivation of the sr protein splicing factor ASF/SF2 results in genomic instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Mischo, H.E.; Gomez-Gonzalez, B.; Grzechnik, P.; Rondon, A.G.; Wei, W.; Steinmetz, L.; Aguilera, A.; Proudfoot, N.J. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell 2011, 41, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Sridhara, S.C.; Carvalho, S.; Grosso, A.R.; Gallego-Paez, L.M.; Carmo-Fonseca, M.; de Almeida, S.F. Transcription dynamics prevent RNA-mediated genomic instability through SRPK2-dependent DDX23 phosphorylation. Cell Rep. 2017, 18, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.C.; Chan, Y.A.; Minaker, S.W.; Aristizabal, M.J.; Barrett, I.; Sipahimalani, P.; Kobor, M.S.; Hieter, P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012, 26, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Drolet, M. Growth inhibition mediated by excess negative supercoiling: The interplay between transcription elongation, R-loop formation and DNA topology. Mol. Microbiol. 2006, 59, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Chedin, F.; Hsieh, C.L.; Wilson, T.E.; Lieber, M.R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gonzalez, B.; Aguilera, A. Activation-Induced Cytidine Deaminase action is strongly stimulated by mutations of the THO complex. Proc. Natl. Acad. Sci. USA 2007, 104, 8409–8414. [Google Scholar] [CrossRef] [PubMed]
- García-Rubio, M.; Barroso, S.; Aguilera, A. Detection of DNA-RNA hybrids in vivo. Methods Mol. Biol. 2017, in press. [Google Scholar]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gomez-Gonzalez, B. DNA-RNA hybrids: The risks of DNA breakage during transcription. Nat. Struct. Mol. Biol. 2017, 24, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Chedin, F. Nascent connections: R-loops and chromatin patterning. Trends Genet. 2016, 32, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.Y.; Stirling, P.C. Replication fork protection factors controlling R-loop bypass and suppression. Genes 2017, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Muse, T.; Aguilera, A. Transcription-replication conflicts: How they occur and how they are resolved. Nat. Rev. 2016, 17, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.C.; Hieter, P. Canonical DNA repair pathways influence R-loop-driven genome instability. J. Mol. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kohler, A.; Hurt, E. Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. 2007, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A. mRNA processing and genomic instability. Nat. Struct. Mol. Biol. 2005, 12, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Klein, H.L. HPR1, a novel yeast gene that prevents intrachromosomal excision recombination, shows carboxy-terminal homology to the Saccharomyces cerevisiae TOP1 gene. Mol. Cell Biol. 1990, 10, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Chavez, S.; Aguilera, A. The yeast HPR1 gene has a functional role in transcriptional elongation that uncovers a novel source of genome instability. Genes Dev. 1997, 11, 3459–3470. [Google Scholar] [CrossRef] [PubMed]
- Chavez, S.; Beilharz, T.; Rondon, A.G.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q.; Lithgow, T.; Aguilera, A. A protein complex containing Tho2, Hpr1, Mft1 and a novel protein, Thp2, connects transcription elongation with mitotic recombination in Saccharomyces cerevisiae. EMBO J. 2000, 19, 5824–5834. [Google Scholar] [CrossRef] [PubMed]
- González-Aguilera, C.; Tous, C.; Gómez-González, B.; Huertas, P.; Luna, R.; Aguilera, A. The THP1-SAC3-SUS1-CDC31 complex works in transcription elongation-mRNA export preventing RNA-mediated genome instability. Mol. Biol. Cell 2008, 19, 4310–4318. [Google Scholar] [CrossRef] [PubMed]
- Piruat, J.I.; Aguilera, A. A novel yeast gene, THO2, is involved in RNA pol II transcription and provides new evidence for transcriptional elongation-associated recombination. EMBO J. 1998, 17, 4859–4872. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Sánchez, M.S.; Barroso, S.; Gómez-González, B.; Luna, R.; Aguilera, A. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011, 7, e1002386. [Google Scholar] [CrossRef] [PubMed]
- Hodroj, D.; Recolin, B.; Serhal, K.; Martínez, S.; Tsanov, N.; Merhi, R.; Maiorano, D. An ATR-dependent function for the DDX19 RNA helicase in nuclear R-loop metabolism. EMBO J. 2017, 36, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Germain, D.R.; Poon, H.Y.; Hildebrandt, M.R.; Monckton, E.A.; McDonald, D.; Hendzel, M.J.; Godbout, R. Dead box 1 facilitates removal of rna and homologous recombination at DNA double-strand breaks. Mol. Cell Biol. 2016, 36, 2794–2810. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.C.; Richard, P.; Patidar, P.L.; Motea, E.A.; Dang, T.T.; Manley, J.L.; Boothman, D.A. XRN2 links transcription termination to DNA damage and replication stress. PLoS Genet. 2016, 12, e1006107. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.A.; Aristizabal, M.J.; Lu, P.Y.; Luo, Z.; Hamza, A.; Kobor, M.S.; Stirling, P.C.; Hieter, P. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet. 2014, 10, e1004288. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chedin, F. Gc skew at the 5’ and 3’ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Nadel, J.; Athanasiadou, R.; Lemetre, C.; Wijetunga, N.A.; Broin, P.O.; Sato, H.; Zhang, Z.; Jeddeloh, J.; Montagna, C.; Golden, A.; et al. RNA:DNA hybrids in the human genome have distinctive nucleotide characteristics, chromatin composition, and transcriptional relationships. Epigenet. Chromatin 2015, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Zeller, P.; Padeken, J.; van Schendel, R.; Kalck, V.; Tijsterman, M.; Gasser, S.M. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 2016, 48, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.T.; Coulson, R.L.; Gonzales, M.L.; Crary, F.K.; Wong, S.S.; Adams, S.; Ach, R.A.; Tsang, P.; Yamada, N.A.; Yasui, D.H.; et al. R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. USA 2013, 110, 13938–13943. [Google Scholar] [CrossRef] [PubMed]
- Taneja, N.; Zofall, M.; Balachandran, V.; Thillainadesan, G.; Sugiyama, T.; Wheeler, D.; Zhou, M.; Grewal, S.I. SNF2 family protein Fft3 suppresses nucleosome turnover to promote epigenetic inheritance and proper replication. Mol. Cell 2017, 66, 50–62.e56. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; McBride, K.M.; Hensley, S.; Lu, Y.; Chedin, F.; Bedford, M.T. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol. Cell 2014, 53, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Nakama, M.; Kawakami, K.; Kajitani, T.; Urano, T.; Murakami, Y. DNA-RNA hybrid formation mediates RNAi-directed heterochromatin formation. Genes Cells 2012, 17, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Pozo, M.; Santos-Pereira, J.M.; Rondón, A.G.; Barroso, S.; Andujar, E.; Pérez-Alegre, M.; García-Muse, T.; Aguilera, A. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 2013, 52, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439. [Google Scholar] [CrossRef] [PubMed]
- El Achkar, E.; Gerbault-Seureau, M.; Muleris, M.; Dutrillaux, B.; Debatisse, M. Premature condensation induces breaks at the interface of early and late replicating chromosome bands bearing common fragile sites. Proc. Natl. Acad. Sci. USA 2005, 102, 18069–18074. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in friedreich ataxia and fragile x syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Loomis, E.W.; Sanz, L.A.; Chedin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.B.; Chen, H.V.; Acharya, D.; Rando, O.J.; Fazzio, T.G. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat. Struct. Mol. Biol. 2015, 22, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gan, H.; Wang, Z.; Lee, J.H.; Zhou, H.; Ordog, T.; Wold, M.S.; Ljungman, M.; Zhang, Z. RPA interacts with HIRA and regulates H3.3 deposition at gene regulatory elements in mammalian cells. Mol. Cell 2017, 65, 272–284. [Google Scholar] [CrossRef] [PubMed]
- García-Pichardo, D.; Cañas, J.C.; García-Rubio, M.L.; Gómez-González, B.; Rondón, A.G.; Aguilera, A. Histone mutants separate R loop formation from genome instability induction. Mol. Cell 2017, 66, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Alzu, A.; Bermejo, R.; Begnis, M.; Lucca, C.; Piccini, D.; Carotenuto, W.; Saponaro, M.; Brambati, A.; Cocito, A.; Foiani, M.; et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 2012, 151, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 2016, 5, e17548. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, R.; Garcia-Rodriguez, N.; Aguilera, A.; Wellinger, R.E. Role for RNA:DNA hybrids in origin-independent replication priming in a eukaryotic system. Proc. Natl. Acad. Sci. USA 2015, 112, 5779–5784. [Google Scholar] [CrossRef] [PubMed]
- Ohle, C.; Tesorero, R.; Schermann, G.; Dobrev, N.; Sinning, I.; Fischer, T. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell 2016, 167, 1001–1013.e7. [Google Scholar] [CrossRef] [PubMed]
- Azvolinsky, A.; Giresi, P.G.; Lieb, J.D.; Zakian, V.A. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 2009, 34, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of replication protein A as a sensor of R loops and a regulator of RNAseH1. Mol. Cell 2017, 65, 832–847.e834. [Google Scholar] [CrossRef] [PubMed]
- Kurat, C.F.; Yeeles, J.T.; Patel, H.; Early, A.; Diffley, J.F. Chromatin controls DNA replication origin selection, lagging-strand synthesis, and replication fork rates. Mol. Cell 2017, 65, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Moyano, E.; Mergui, X.; Garcia-Rubio, M.L.; Barroso, S.; Aguilera, A. The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014, 28, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.D.; D’Andrea, A.D. The Fanconi Anemia/BRCA pathway: New faces in the crowd. Genes Dev. 2005, 19, 2925–2940. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi anemia pathway protects genome integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.L.T.; Pohl, T.J.; Chen, C.F.; Chan, A.; Pott, S.; Zakian, V.A. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat. Commun. 2017, 8, 15025. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat. Rev. 2004, 4, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Zhang, Z.; Lu, Z.; Hsieh, C.L.; Lieber, M.R. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: A nick can serve as a strong R-loop initiation site. Mol. Cell Biol. 2010, 30, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J.; Rolland, T.; Adelmant, G.; Xia, X.; Owen, M.S.; Dricot, A.; Zack, T.I.; Sahni, N.; Jacob, Y.; Hao, T.; et al. Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev. 2014, 28, 1957–1975. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Cipak, L.; Watanabe, N.; Bessho, T. The role of BRCA2 in replication-coupled DNA interstrand cross-link repair in vitro. Nat. Struct. Mol. Biol. 2006, 13, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Lomonosov, M.; Anand, S.; Sangrithi, M.; Davies, R.; Venkitaraman, A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003, 17, 3017–3022. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.J.; Wang, Y.; Lee, M.; Venkitaraman, A.R. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science 2004, 306, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Mondal, G.; Rowley, M.; Guidugli, L.; Wu, J.; Pankratz, V.S.; Couch, F.J. BRCA2 localization to the midbody by filamin a regulates CEP55 signaling and completion of cytokinesis. Dev. Cell 2012, 23, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zou, C.; Bai, Y.; Wazer, D.E.; Band, V.; Gao, Q. DSS1 is required for the stability of BRCA2. Oncogene 2006, 25, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.J.; Joenje, H. Fanconi anemia and DNA replication repair. DNA Repair 2007, 6, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Lossaint, G.; Larroque, M.; Ribeyre, C.; Bec, N.; Larroque, C.; Decaillet, C.; Gari, K.; Constantinou, A. FANCD2 binds MCM proteins and controls replisome function upon activation of S phase checkpoint signaling. Mol. Cell 2013, 51, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Andreassen, P.R.; D’Andrea, A.D. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol. Cell. Biol. 2004, 24, 5850–5862. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Sanz, L.A.; Xu, X.; Hartono, S.R.; Chedin, F. Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutieres syndrome. Elife 2015, 4, e08007. [Google Scholar] [CrossRef] [PubMed]
- Sagie, S.; Toubiana, S.; Hartono, S.R.; Katzir, H.; Tzur-Gilat, A.; Havazelet, S.; Francastel, C.; Velasco, G.; Chedin, F.; Selig, S. Telomeres in ICF syndrome cells are vulnerable to DNA damage due to elevated DNA:RNA hybrids. Nat. Commun. 2017, 8, 14015. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatia, V.; Herrera-Moyano, E.; Aguilera, A.; Gómez-González, B. The Role of Replication-Associated Repair Factors on R-Loops. Genes 2017, 8, 171. https://doi.org/10.3390/genes8070171
Bhatia V, Herrera-Moyano E, Aguilera A, Gómez-González B. The Role of Replication-Associated Repair Factors on R-Loops. Genes. 2017; 8(7):171. https://doi.org/10.3390/genes8070171
Chicago/Turabian StyleBhatia, Vaibhav, Emilia Herrera-Moyano, Andrés Aguilera, and Belén Gómez-González. 2017. "The Role of Replication-Associated Repair Factors on R-Loops" Genes 8, no. 7: 171. https://doi.org/10.3390/genes8070171
APA StyleBhatia, V., Herrera-Moyano, E., Aguilera, A., & Gómez-González, B. (2017). The Role of Replication-Associated Repair Factors on R-Loops. Genes, 8(7), 171. https://doi.org/10.3390/genes8070171