
Pre-Replicative Repair of Oxidized Bases Maintains Fidelity in Mammalian Genomes: The Cowcatcher Role of NEIL1 DNA Glycosylase



Abstract
:1. Introduction
2. Key Features of Oxidative Damage Repair in the Mammalian Genome
2.1. A Plethora of Oxidized Bases and Their Repair: A Constant Challenge for the Mammalian
2.2. Minimal BER Reaction
3. Complex Regulation of Mammalian Base Excision Repair
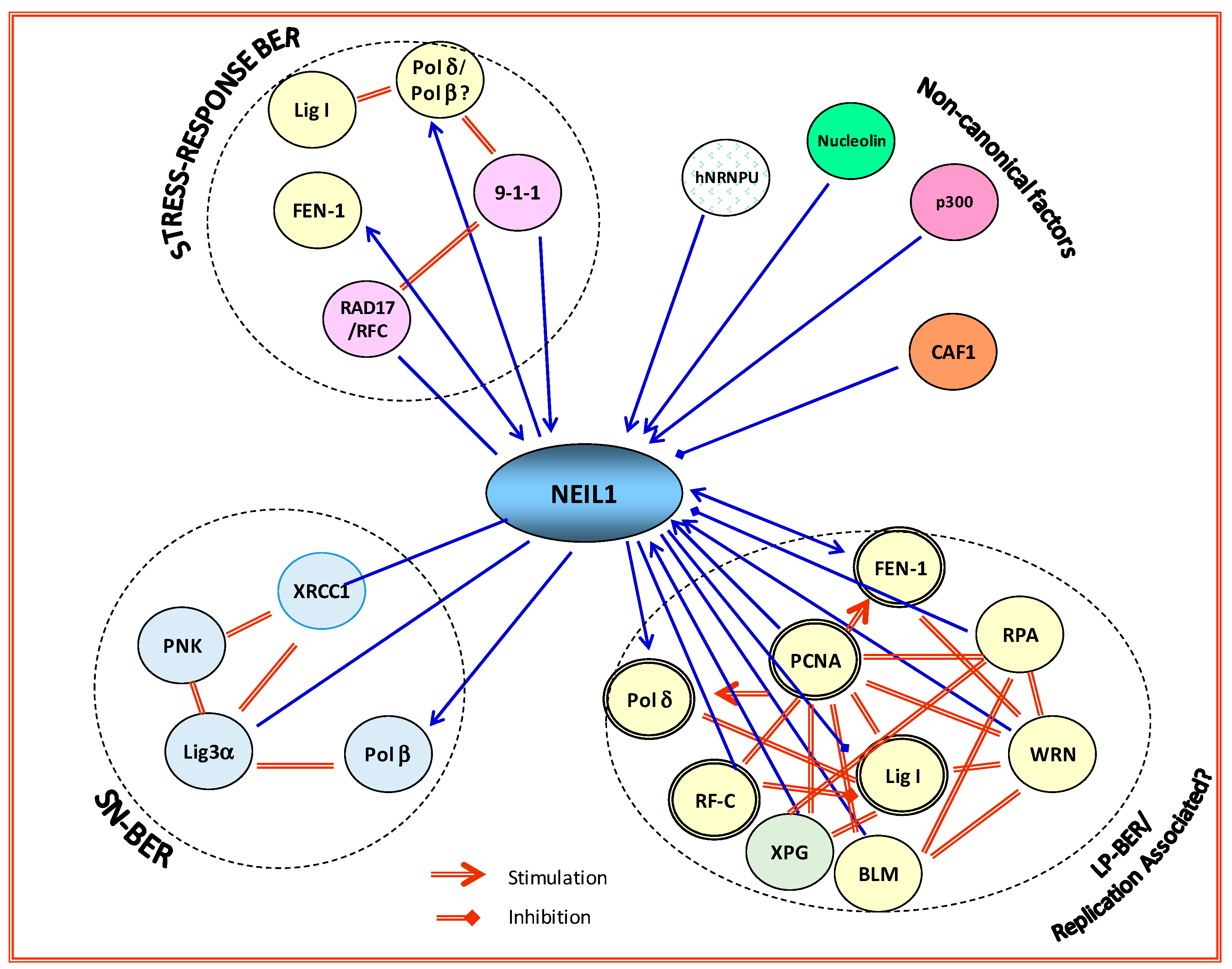
3.1. Preformed BERosome Complexes Regulated by Multiple Posttranslational Modifications
3.2. DNA Glycosilases May Direct Base Excision Repair Sub-Pathway Choice via Specific Interaction with Downstream Repair Proteins
3.3. Individual Dispensability and Overlapping Substrate Specificity of DNA Glycosylases
4. Replicating and Transcribing DNA Employ Distinct Base Excision Repair Sub-Pathways
4.1. Replication-Associated Base Excision Repair Is Critical for Preventing Mutations in Cycling Cells
4.2. Pre-Replicative vs. Post-Replicative Base Excision Repair
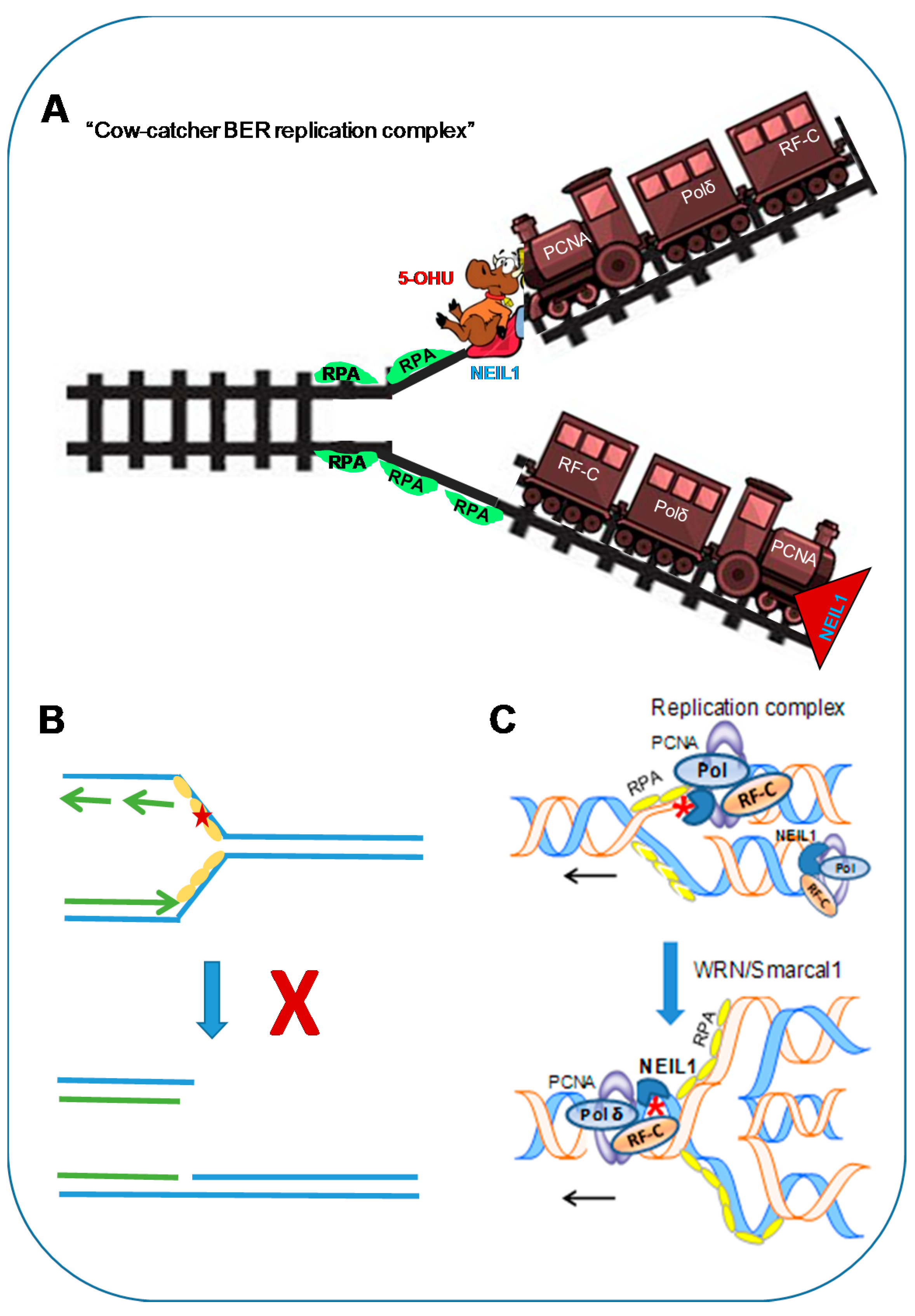
5. Cowcatcher Model of Pre-Replicative Repair: Molecular Insights into Template Strand Repair at the Replication Fork
5.1. Damage Recognition in single-stranded DNA Template Strand to Stall Replication
5.2. Pre-Replicative Repair in the Re-Annealed Duplex in the Regressed Fork Structure
5.3. Backup Function of NEIL2 in Pre-Replicative BER
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
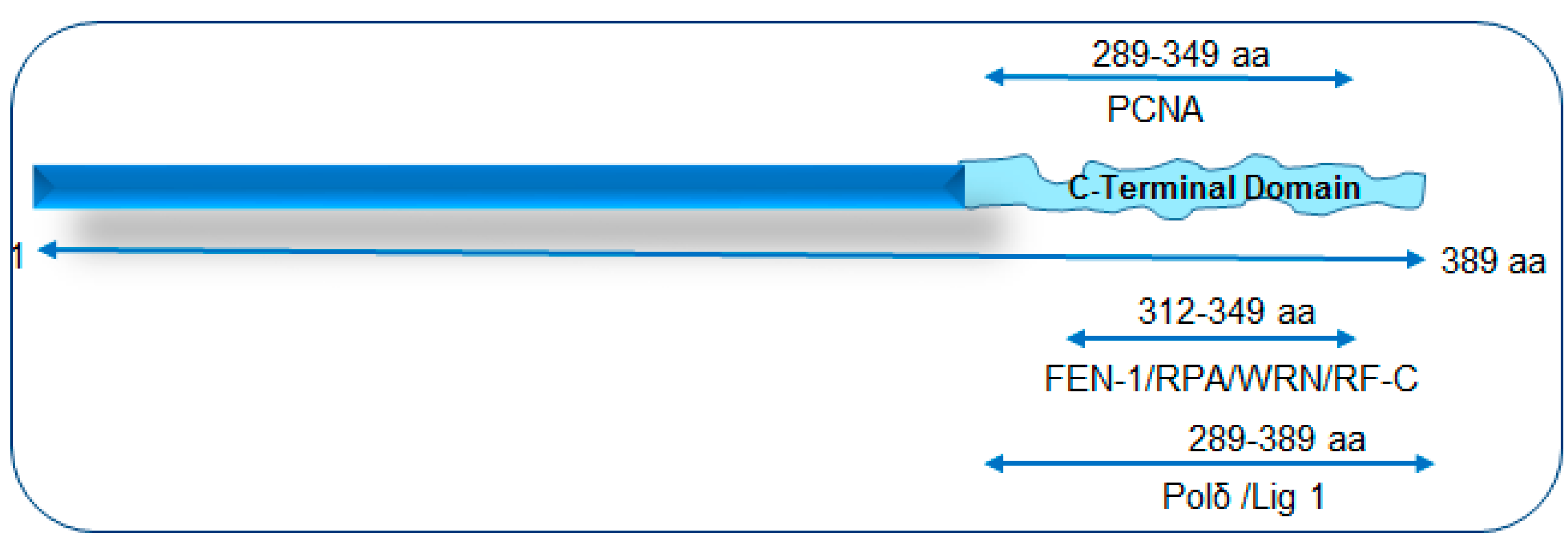
- Hegde, P.M.; Dutta, A.; Sengupta, S.; Mitra, J.; Adhikari, S.; Tomkinson, A.E.; Li, G.M.; Boldogh, I.; Hazra, T.K.; Mitra, S.; et al. The C-terminal Domain (CTD) of Human DNA Glycosylase NEIL1 Is Required for Forming BERosome Repair Complex with DNA Replication Proteins at the Replicating Genome: DOMINANT NEGATIVE FUNCTION OF THE CTD. J. Biol. Chem. 2015, 290, 20919–20933. [Google Scholar] [CrossRef] [PubMed]
- Bjelland, S.; Seeberg, E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 2003, 531, 37–80. [Google Scholar] [CrossRef] [PubMed]
- Kreutzer, D.A.; Essigmann, J.M. Oxidized, deaminated cytosines are a source of C --> T transitions in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 3578–3582. [Google Scholar] [CrossRef] [PubMed]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013, 3, 120144. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.L.; Rangaswamy, S.; Wicker, C.A.; Izumi, T. Repair of oxidative DNA damage and cancer: Recent progress in DNA base excision repair. Antioxid. Redox Signal. 2014, 20, 708–726. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Aguilera, A.; Gellert, M.; Hanawalt, P.C.; Hays, J.B.; Lehmann, A.R.; Lindahl, T.; Lowndes, N.; Sarasin, A.; Wood, R.D. DNA repair: From molecular mechanism to human disease. DNA Repair 2006, 5, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G.L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 2005, 434, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Cadet, J.; Moller, L.; Poulsen, H.E.; Vina, J. Are we sure we know how to measure 8-oxo-7,8-dihydroguanine in DNA from human cells? Arch. Biochem. Biophys. 2004, 423, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Aboujaoude, E.N.; Chen, Q.; Ames, B.N. Assays of oxidative DNA damage biomarkers 8-oxo-2′-deoxyguanosine and 8-oxoguanine in nuclear DNA and biological fluids by high-performance liquid chromatography with electrochemical detection. Methods Enzymol. 1994, 234, 16–33. [Google Scholar] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Banerjee, S.; Hegde, P.M.; Bellot, L.J.; Hazra, T.K.; Boldogh, I.; Mitra, S. Enhancement of NEIL1 protein-initiated oxidized DNA base excision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via direct interaction. J. Biol. Chem. 2012, 287, 34202–34211. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Functions of disordered regions in mammalian early base excision repair proteins. Cell. Mol. Life Sci. 2010, 67, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, X.; Lu, J.; Liang, H.; Dai, Q.; Xu, G.L.; Luo, C.; Jiang, H.; He, C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012, 8, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Drablos, F.; Slupphaug, G. Uracil in DNA--occurrence, consequences and repair. Oncogene 2002, 21, 8935–8948. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.L.; Li, X.; Gu, Y.; Wright, P.M.; Chang, D.Y. Repair of oxidative DNA damage: Mechanisms and functions. Cell Biochem. Biophys. 2001, 35, 141–170. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, V.; Sunkara, S.; Wallace, S.S.; Bond, J.P. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair 2002, 1, 517–529. [Google Scholar] [CrossRef]
- Burrows, C.J.; Muller, J.G.; Kornyushyna, O.; Luo, W.; Duarte, V.; Leipold, M.D.; David, S.S. Structure and potential mutagenicity of new hydantoin products from guanosine and 8-oxo-7,8-dihydroguanine oxidation by transition metals. Environ. Health Perspect. 2002, 110, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Bailly, V.; Verly, W.G. Escherichia coli endonuclease III is not an endonuclease but a β-elimination catalyst. Biochem. J. 1987, 242, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O.; Golan, G.; Gilboa, R.; Fernandes, A.S.; Gerchman, S.E.; Kycia, J.H.; Rieger, R.A.; Grollman, A.P.; Shoham, G. Structural analysis of an Escherichia coli endonuclease VIII covalent reaction intermediate. EMBO J. 2002, 21, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Frosina, G.; Fortini, P.; Rossi, O.; Carrozzino, F.; Raspaglio, G.; Cox, L.S.; Lane, D.P.; Abbondandolo, A.; Dogliotti, E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996, 271, 9573–9578. [Google Scholar] [CrossRef] [PubMed]
- Podlutsky, A.J.; Dianova, I.I.; Podust, V.N.; Bohr, V.A.; Dianov, G.L. Human DNA polymerase β initiates DNA synthesis during long-patch repair of reduced AP sites in DNA. EMBO J. 2001, 20, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Busso, C.S.; Lake, M.W.; Izumi, T. Posttranslational modification of mammalian AP endonuclease (APE1). Cell. Mol. Life Sci. 2010, 67, 3609–3620. [Google Scholar] [CrossRef] [PubMed]
- Seet, B.T.; Dikic, I.; Zhou, M.M.; Pawson, T. Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell. Biol. 2006, 7, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.Y.C.; Hegde, M.L.; Hegde, P.M.; Mitra, J.; Pandey, A.; Dutta, A.; Datarwala, A.D.; Bhakat, K.K.; Mitra, S. Acetylation of NEIL1 is required for its repair complex formation in chromatin: Associated with resistance to oxidative stress. J. Biol. Chem. 2017, in press. [Google Scholar]
- Bhakat, K.K.; Hazra, T.K.; Mitra, S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004, 32, 3033–3039. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Cao, V.B.; Doublie, S. Phosphorylation Sites Identified in the NEIL1 DNA Glycosylase Are Potential Targets for the JNK1 Kinase. PLoS ONE 2016, 11, e0157860. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Roy, R. Truncation of amino-terminal tail stimulates activity of human endonuclease III (hNTH1). J. Mol. Biol. 2002, 321, 265–276. [Google Scholar] [CrossRef]
- Nilsen, H.; Otterlei, M.; Haug, T.; Solum, K.; Nagelhus, T.A.; Skorpen, F.; Krokan, H.E. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997, 25, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Bhakat, K.K.; Izumi, T.; Yang, S.H.; Hazra, T.K.; Mitra, S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003, 22, 6299–6309. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hegde, P.M.; Arijit, D.; Boldogh, I.; Mitra, S. Human DNA Glycosylase NEIL1’s Interactions with Downstream Repair Proteins Is Critical for Efficient Repair of Oxidized DNA Base Damage and Enhanced Cell Survival. Biomolecules 2012, 2, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008, 283, 3130–3140. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Moharana, K.; Wallace, S.S.; Doublie, S. Destabilization of the PCNA trimer mediated by its interaction with the NEIL1 DNA glycosylase. Nucleic Acids Res. 2017, 45, 2897–2909. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Dyrkheeva, N.; Williams, J.; Wilson, S.H. Mammalian Base Excision Repair: Functional Partnership between PARP-1 and APE1 in AP-Site Repair. PLoS ONE 2015, 10, e0124269. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Tsutakawa, S.E.; Hegde, P.M.; Holthauzen, L.M.; Li, J.; Oezguen, N.; Hilser, V.J.; Tainer, J.A.; Mitra, S. The disordered C-terminal domain of human DNA glycosylase NEIL1 contributes to its stability via intramolecular interactions. J. Mol. Biol. 2013, 425, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Tann, A.W.; Longley, M.J.; Copeland, W.C.; Mitra, S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008, 283, 26349–26356. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.H.; Kunkel, T.A. Passing the baton in base excision repair. Nat. Struct. Biol. 2000, 7, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sengupta, S.; Hegde, P.M.; Mitra, J.; Jiang, S.; Holey, B.; Sarker, A.H.; Tsai, M.S.; Hegde, M.L.; Mitra, S. Regulation of oxidized base damage repair by chromatin assembly factor 1 subunit A. Nucleic Acids Res. 2017, 45, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Bohr, V.A.; Smith, C.A.; Okumoto, D.S.; Hanawalt, P.C. DNA repair in an active gene: Removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 1985, 40, 359–369. [Google Scholar] [CrossRef]
- Dutta, A.; Yang, C.; Sengupta, S.; Mitra, S.; Hegde, M.L. New paradigms in the repair of oxidative damage in human genome: Mechanisms ensuring repair of mutagenic base lesions during replication and involvement of accessory proteins. Cell. Mol. Life Sci. 2015, 72, 1679–1698. [Google Scholar] [CrossRef] [PubMed]
- Kadyrov, F.A.; Holmes, S.F.; Arana, M.E.; Lukianova, O.A.; O’Donnell, M.; Kunkel, T.A.; Modrich, P. Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J. Biol. Chem. 2007, 282, 37181–37190. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Wakamiya, M.; Venkova-Canova, T.; Pandita, R.K.; Aguilera-Aguirre, L.; Sarker, A.H.; Singh, D.K.; Hosoki, K.; Wood, T.G.; Sharma, G.; et al. Neil2-null Mice Accumulate Oxidized DNA Bases in the Transcriptionally Active Sequences of the Genome and Are Susceptible to Innate Inflammation. J. Biol. Chem. 2015, 290, 24636–24648. [Google Scholar] [CrossRef] [PubMed]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D.E. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13300–13305. [Google Scholar] [CrossRef] [PubMed]
- Minowa, O.; Arai, T.; Hirano, M.; Monden, Y.; Nakai, S.; Fukuda, M.; Itoh, M.; Takano, H.; Hippou, Y.; Aburatani, H.; et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4156–4161. [Google Scholar] [CrossRef] [PubMed]
- Osterod, M.; Hollenbach, S.; Hengstler, J.G.; Barnes, D.E.; Lindahl, T.; Epe, B. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis 2001, 22, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Marth, J.D.; Orban, P.C.; Mossmann, H.; Rajewsky, K. Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science 1994, 265, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Carroll, B.L.; Sweasy, J.B.; Wallace, S.S.; Doublie, S. Genome and cancer single nucleotide polymorphisms of the human NEIL1 DNA glycosylase: Activity, structure, and the effect of editing. DNA Repair 2014, 14, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Tao, H.; Goto, M.; Igarashi, H.; Taniguchi, T.; Maekawa, M.; Takezaki, T.; Sugimura, H. Inactivating mutations of the human base excision repair gene NEIL1 in gastric cancer. Carcinogenesis 2004, 25, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Broderick, P.; Bagratuni, T.; Vijayakrishnan, J.; Lubbe, S.; Chandler, I.; Houlston, R.S. Evaluation of NTHL1, NEIL1, NEIL2, MPG, TDG, UNG and SMUG1 genes in familial colorectal cancer predisposition. BMC Cancer 2006, 6, 243. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Shinmura, K.; Tao, H.; Tsugane, S.; Sugimura, H. Three novel NEIL1 promoter polymorphisms in gastric cancer patients. World J. Gastrointest. Oncol. 2010, 2, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Kato, H.; Kawanishi, Y.; Igarashi, H.; Goto, M.; Tao, H.; Inoue, Y.; Nakamura, S.; Misawa, K.; Mineta, H.; et al. Abnormal Expressions of DNA Glycosylase Genes NEIL1, NEIL2, and NEIL3 Are Associated with Somatic Mutation Loads in Human Cancer. Oxid. Med. Cell. Longev. 2016, 2016, 1546392. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Zhao, H.; Liu, Z.; Wang, L.E.; El-Naggar, A.K.; Sturgis, E.M.; Wei, Q. Functional variants of the NEIL1 and NEIL2 genes and risk and progression of squamous cell carcinoma of the oral cavity and oropharynx. Clin. Cancer Res. 2008, 14, 4345–4352. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Kwiatkowski, D.; Galecki, P.; Talarowska, M.; Orzechowska, A.; Bobinska, K.; Bielecka-Kowalska, A.; Szemraj, J.; Maes, M.; Su, K.P.; et al. Association between single nucleotide polymorphisms of MUTYH, hOGG1 and NEIL1 genes, and depression. J. Affect. Disord. 2015, 184, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, M.; Zhang, Z.; Jiang, G.; Fu, X.; Fan, M.; Sun, M.; Wei, Q.; Zhao, K. A NEIL1 single nucleotide polymorphism (rs4462560) predicts the risk of radiation-induced toxicities in esophageal cancer patients treated with definitive radiotherapy. Cancer 2013, 119, 4205–4211. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc. Natl. Acad. Sci. USA 1974, 71, 3649–3653. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Kelly, V.P.; Minowa, O.; Noda, T.; Nishimura, S. The study using wild-type and Ogg1 knockout mice exposed to potassium bromate shows no tumor induction despite an extensive accumulation of 8-hydroxyguanine in kidney DNA. Toxicology 2006, 221, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.K.; Ocampo-Hafalla, M.T.; Vartanian, V.; Jaruga, P.; Kirkali, G.; Koenig, K.L.; Brown, S.; Lloyd, R.S.; Dizdaroglu, M.; Teebor, G.W. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair 2009, 8, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Zhao, X.; Burrows, C.J.; David, S.S. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry 2008, 47, 7137–7146. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.B.; Bianchet, M.A.; Krosky, D.J.; Friedman, J.I.; Amzel, L.M.; Stivers, J.T. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature 2007, 449, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Slupphaug, G.; Mol, C.D.; Kavli, B.; Arvai, A.S.; Krokan, H.E.; Tainer, J.A. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature 1996, 384, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Izumi, T.; Boldogh, I.; Bhakat, K.K.; Hill, J.W.; Hazra, T.K. Choreography of oxidative damage repair in mammalian genomes. Free Radic. Biol. Med. 2002, 33, 15–28. [Google Scholar] [CrossRef]
- Osorio, A.; Milne, R.L.; Kuchenbaecker, K.; Vaclova, T.; Pita, G.; Alonso, R.; Peterlongo, P.; Blanco, I.; de la Hoya, M.; Duran, M.; et al. DNA glycosylases involved in base excision repair may be associated with cancer risk in BRCA1 and BRCA2 mutation carriers. PLoS Genet. 2014, 10, e1004256. [Google Scholar] [CrossRef] [PubMed]
- Elingarami, S.; Liu, H.; Kalinjuma, A.V.; Hu, W.; Li, S.; He, N. Polymorphisms in NEIL-2, APE-1, CYP2E1 and MDM2 Genes are Independent Predictors of Gastric Cancer Risk in a Northern Jiangsu Population (China). J. Nanosci. Nanotechnol. 2015, 15, 4815–4828. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Sunter, N.J.; Singh, P.B.; Austin, C.A.; Durkacz, B.W.; Tilby, M.J. gammaH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS ONE 2007, 2, e1057. [Google Scholar] [CrossRef] [PubMed]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin compaction protects genomic DNA from radiation damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef] [PubMed]
- Berquist, B.R.; Wilson, D.M., 3rd. Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett. 2012, 327, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Odell, I.D.; Wallace, S.S.; Pederson, D.S. Rules of engagement for base excision repair in chromatin. J. Cell. Physiol. 2013, 228, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 2011, 286, 6006–6016. [Google Scholar] [CrossRef] [PubMed]
- Parlanti, E.; Locatelli, G.; Maga, G.; Dogliotti, E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007, 35, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. Formation and processing of DNA damage substrates for the hNEIL enzymes. Free Radic. Biol. Med. 2017, 107, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fleming, A.M.; Averill, A.M.; Burrows, C.J.; Wallace, S.S. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015, 43, 4039–4054. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.A.; Hegde, M.L.; Hazra, T.K.; Mitra, S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair 2010, 9, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Theriot, C.A.; Das, A.; Hegde, P.M.; Guo, Z.; Gary, R.K.; Hazra, T.K.; Shen, B.; Mitra, S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 2008, 283, 27028–27037. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Boldogh, I.; Lee, J.W.; Harrigan, J.A.; Hegde, M.L.; Piotrowski, J.; de Souza Pinto, N.; Ramos, W.; Greenberg, M.M.; Hazra, T.K.; et al. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J. Biol. Chem. 2007, 282, 26591–26602. [Google Scholar] [CrossRef] [PubMed]
- Aladjem, M.I. Replication in context: Dynamic regulation of DNA replication patterns in metazoans. Nat. Rev. Genet. 2007, 8, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Rampakakis, E.; Arvanitis, D.N.; Di Paola, D.; Zannis-Hadjopoulos, M. Metazoan origins of DNA replication: Regulation through dynamic chromatin structure. J. Cell. Biochem. 2009, 106, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, R.A.; Holzen, T.M. Cell cycle regulation of DNA replication. Annu. Rev. Genet. 2007, 41, 237–280. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.P.; Dutta, A. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 2002, 71, 333–374. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Bell, S.P. Initiation of DNA replication in eukaryotic cells. Annu. Rev. Cell Dev. Biol. 1997, 13, 293–332. [Google Scholar] [CrossRef] [PubMed]
- Hook, S.S.; Lin, J.J.; Dutta, A. Mechanisms to control rereplication and implications for cancer. Curr. Opin. Cell Biol. 2007, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Krasinska, L.; Besnard, E.; Cot, E.; Dohet, C.; Mechali, M.; Lemaitre, J.M.; Fisher, D. Cdk1 and Cdk2 activity levels determine the efficiency of replication origin firing in Xenopus. EMBO J. 2008, 27, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Wilson, D.M., 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Solvang-Garten, K.; Hanssen-Bauer, A.; Lieske, N.V.; Pettersen, H.S.; Pettersen, G.K.; Wilson, D.M., 3rd; Krokan, H.E.; Otterlei, M. Direct interaction between XRCC1 and UNG2 facilitates rapid repair of uracil in DNA by XRCC1 complexes. DNA Repair 2010, 9, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Otterlei, M.; Warbrick, E.; Nagelhus, T.A.; Haug, T.; Slupphaug, G.; Akbari, M.; Aas, P.A.; Steinsbekk, K.; Bakke, O.; Krokan, H.E. Post-replicative base excision repair in replication foci. EMBO J. 1999, 18, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y. Molecular mechanism of PCNA-dependent base excision repair. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 68, 129–138. [Google Scholar] [PubMed]
- Jiricny, J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012633. [Google Scholar] [CrossRef] [PubMed]
- Bjoras, K.O.; Sousa, M.M.L.; Sharma, A.; Fonseca, D.M.; Sogaard, C.K.; Bjoras, M.; Otterlei, M. Monitoring of the spatial and temporal dynamics of BER/SSBR pathway proteins, including MYH, UNG2, MPG, NTH1 and NEIL1-3, during DNA replication. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hazra, T.K.; Izumi, T.; Maidt, L.; Floyd, R.A.; Mitra, S. The presence of two distinct 8-oxoguanine repair enzymes in human cells: Their potential complementary roles in preventing mutation. Nucleic Acids Res. 1998, 26, 5116–5122. [Google Scholar] [CrossRef] [PubMed]
- Popuri, V.; Croteau, D.L.; Bohr, V.A. Substrate specific stimulation of NEIL1 by WRN but not the other human RecQ helicases. DNA Repair 2010, 9, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Postow, L.; Woo, E.M.; Chait, B.T.; Funabiki, H. Identification of SMARCAL1 as a component of the DNA damage response. J. Biol. Chem. 2009, 284, 35951–35961. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, R.; Cimprich, K.A. HARPing on about the DNA damage response during replication. Genes Dev. 2009, 23, 2359–2365. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Mitra, S.; Houston Methodist Research Institute, Houston, TX, USA. Unpublished Work. 2017.
- Bloom, L.B. Loading clamps for DNA replication and repair. DNA Repair 2009, 8, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Parrilla-Castellar, E.R.; Arlander, S.J.; Karnitz, L. Dial 9-1-1 for DNA damage: The Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair 2004, 3, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Gasser, S.M.; Shimada, K. Replication checkpoint: Tuning and coordination of replication forks in s phase. Genes 2013, 4, 388–434. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, V.P.; Lindsey-Boltz, L.A.; Cesare, A.J.; Maniwa, Y.; Griffith, J.D.; Hurwitz, J.; Sancar, A. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Brandt, P.D.; Lindsey-Boltz, L.A.; Sancar, A.; Bambara, R.A. Long patch base excision repair proceeds via coordinated stimulation of the multienzyme DNA repair complex. J. Biol. Chem. 2009, 284, 15158–15172. [Google Scholar] [CrossRef] [PubMed]
- Neurauter, C.G.; Luna, L.; Bjoras, M. Release from quiescence stimulates the expression of human NEIL3 under the control of the Ras dependent ERK-MAP kinase pathway. DNA Repair 2012, 11, 401–409. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mammalian DG | Preferred Substrates |
|---|---|
| NEIL1 | FapyA, FapyG, Tg, 5-OHC, 5-OHU, DHU, 5-formyl U, DHU, DHT, single stranded 8-oxo-G (oxo-G opposite C), hydantoin lesions, Gh, Sp, 5OHMH |
| NEIL2 | 5-OHC, 5-OHU, DHT, DHU, Tg, 8-oxo-G, Gh, Sp |
| NEIL3 | FapyA, FapyG, Tg, 5-OHC, 5-OHU, DHU, DHT, Gh, Sp, 5OHMH |
| OGG1 | 8-oxo-G, 8-Oxo-A, Fapy G; prefers lesion opposite C |
| NTHL1 | 5-OHU, 5-OHC, TG, DHU, and FapyG |
| Gene | SNP Database Entry Number | Reported Risks | References |
|---|---|---|---|
| NEIL1 | rs4462560 | Radiation-induced esophageal toxicity, depression disorders. | [60,61] |
| rs5745908 | Familial colorectal cancer. | [56] | |
| NEIL2 | rs804269, rs804268, rs8191613, rs8191642, rs8191663, rs8191664, rs1534862, rs8191667 | Familial colorectal cancer. | [56] |
| rs146678 | Associated with breast cancer risk in BRCA2 mutation carriers. | [70] | |
| rs804270 | Increased risk for gastric cancer, increased risk of squamous cell carcinomas of the oral cavity and oropharynx. | [59,71] |
| DNA Replication Proteins | Functional Association with NEIL1 | Reference |
|---|---|---|
| PCNA | PCNA stimulates NEIL1 activity in excising 5-OHU from single-stranded DNA sequences, including fork structures. PCNA enhances NEIL1 loading on the substrate. | [36,38,39,82] |
| FEN-1 | NEIL1 participates in strand displacement repair synthesis (LP-BER) mediated by FEN-1 and stimulated by PCNA. FEN-1 cleaves the 5′-overhanging flap structure that is generated by displacement synthesis when DNA polymerase encounters the 5′ end of a downstream Okazaki fragment. | [82] |
| RPA | RPA coats the ssDNA template at the replication fork and inhibits NEIL1’s activity (to regulate excision of oxidative DNA base damage in primer-template structures) via direct interaction, as shown through in vivo and in vitro analysis. | [81] |
| RF-C | RF-C activates NEIL1-initiated LP-BER along with DNA replication proteins as shown through in vivo and in vitro analysis. | [3] |
| Polδ | NEIL1 physically interacts with Polδ as shown by in vivo and in vitro analysis. | [3] |
| Lig 1 | NEIL1 physically interacts with Lig 1 as shown by in vivo and in vitro analysis. | [3] |
| WRN | WRN stimulates NEIL1 to excise oxidative lesions from bubble DNA substrates. | [83] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangaswamy, S.; Pandey, A.; Mitra, S.; Hegde, M.L. Pre-Replicative Repair of Oxidized Bases Maintains Fidelity in Mammalian Genomes: The Cowcatcher Role of NEIL1 DNA Glycosylase. Genes 2017, 8, 175. https://doi.org/10.3390/genes8070175
Rangaswamy S, Pandey A, Mitra S, Hegde ML. Pre-Replicative Repair of Oxidized Bases Maintains Fidelity in Mammalian Genomes: The Cowcatcher Role of NEIL1 DNA Glycosylase. Genes. 2017; 8(7):175. https://doi.org/10.3390/genes8070175
Chicago/Turabian StyleRangaswamy, Suganya, Arvind Pandey, Sankar Mitra, and Muralidhar L. Hegde. 2017. "Pre-Replicative Repair of Oxidized Bases Maintains Fidelity in Mammalian Genomes: The Cowcatcher Role of NEIL1 DNA Glycosylase" Genes 8, no. 7: 175. https://doi.org/10.3390/genes8070175
APA StyleRangaswamy, S., Pandey, A., Mitra, S., & Hegde, M. L. (2017). Pre-Replicative Repair of Oxidized Bases Maintains Fidelity in Mammalian Genomes: The Cowcatcher Role of NEIL1 DNA Glycosylase. Genes, 8(7), 175. https://doi.org/10.3390/genes8070175