
Zebrafish Xenograft: An Evolutionary Experiment in Tumour Biology



Abstract
:1. Introduction
2. Cancer Context
2.1. Structure of the Tumour Microenvironment
2.2. Migration and Invasion
2.3. Metastasis: Beyond Migration and Invasion
3. Cancer out of Context
3.1. Simulating the Microenvironment
3.2. The Immune Problem
4. Zebrafish Xenograft: An Evolutionary Experiment
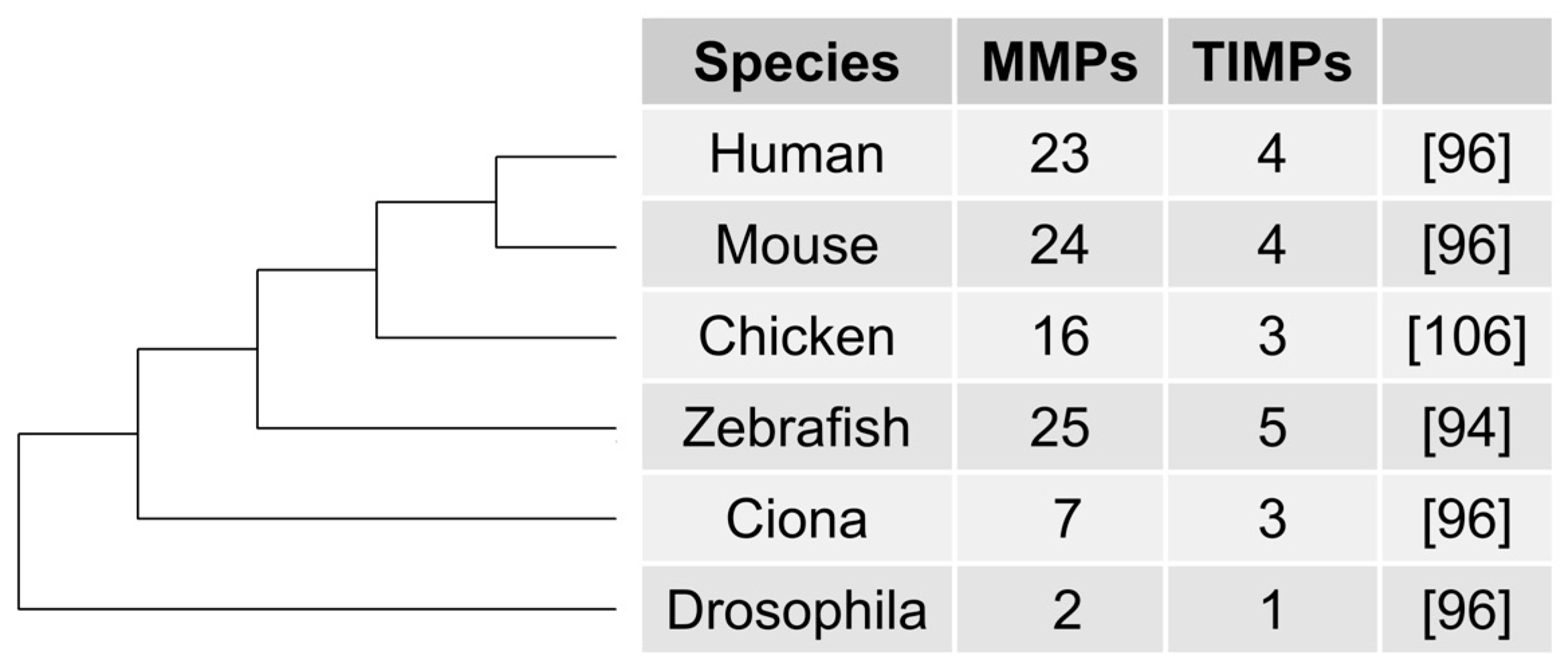
4.1. Case Study: Matrix Metalloproteinases
4.2. Xenografting as an Evolutionary Experiment
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, R.; Curry, J.M.; Roy, L.D.; Grover, P.; Haider, J.; Moore, L.J.; Wu, S.-T.; Kamesh, A.; Yazdanifar, M.; Ahrens, W.A.; et al. A novel association of neuropilin-1 and MUC1 in pancreatic ductal adenocarcinoma: Role in induction of VEGF signaling and angiogenesis. Oncogene 2016, 35, 5608–5618. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Tang, S.-C.; Cai, Y.; Pi, W.; Deng, L.; Wu, G.; Chavanieu, A.; Teng, Y. Suppression of breast cancer metastasis through the inactivation of ADP-ribosylation factor 1. Oncotarget 2016, 7, 58111–58120. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Xie, X.; Walker, S.; White, D.T.; Mumm, J.S.; Cowell, J.K. Evaluating human cancer cell metastasis in zebrafish. BMC Cancer 2013, 13, 453. [Google Scholar] [CrossRef] [PubMed]
- Wertman, J.; Veinotte, C.J.; Dellaire, G.; Berman, J.N. The zebrafish xenograft platform: Evolution of a novel cancer model and preclinical screening tool. Adv. Exp. Med. Biol. 2016, 916, 289–314. [Google Scholar] [CrossRef] [PubMed]
- Tulotta, C.; He, S.; Chen, L.; Groenewoud, A.; van der Ent, W.; Meijer, A.; Spaink, H.; Snaar-Jagalska, B.E. Imaging of human cancer cell proliferation, invasion, and micrometastasis in a zebrafish xenogeneic engraftment model. In Zebrafish; Kawakami, K., Patton, E.E., Orger, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 155–169. ISBN 978-1-4939-3769-1. [Google Scholar]
- Ignatius, M.S.; Hayes, M.; Langenau, D.M. In Vivo imaging of cancer in zebrafish. Adv. Exp. Med. Biol. 2016, 916, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; Snaar-Jagalska, B.E.; Ten Dijke, P. Fish tales: The use of zebrafish xenograft human cancer cell models. Histol. Histopathol. 2017, 32, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.M.; Santos, F.C.; Dingli, D. The ecology of cancer from an evolutionary game theory perspective. Interface Focus 2014, 4, 20140019. [Google Scholar] [CrossRef] [PubMed]
- Korolev, K.S.; Xavier, J.B.; Gore, J. Turning ecology and evolution against cancer. Nat. Rev. Cancer 2014, 14, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-I.; Wang, H.-Y.; Ling, S.; Lu, X. The ecology and evolution of cancer: The ultra-microevolutionary process. Annu. Rev. Genet. 2016, 50, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Orlando, P.A.; Gatenby, R.A.; Brown, J.S. Tumor evolution in space: The effects of competition colonization tradeoffs on tumor invasion dynamics. Front. Oncol. 2013, 3, 45. [Google Scholar] [CrossRef] [PubMed]
- Torkamani, A.; Schork, N.J. Identification of rare cancer driver mutations by network reconstruction. Genome Res. 2009, 19, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J. Cancer drug discovery and anticancer drug development. In The Molecular Basis of Human Cancer; Humana Press: New York, NY, USA, 2017; pp. 695–707. ISBN 978-1-934115-18-3. [Google Scholar]
- Newell, H.; Sausville, E. Cytotoxic drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El-Rayes, B.; Ribatti, D.; et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35, S224–S243. [Google Scholar] [CrossRef] [PubMed]
- Stock, A.-M.; Troost, G.; Niggemann, B.; Zänker, K.S.; Entschladen, F. Targets for anti-metastatic drug development. Curr. Pharm. Des. 2013, 19, 5127–5134. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Combining cancer immunotherapy and targeted therapy. Curr. Opin. Immunol. 2013, 25, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Sakai, A.K.; Allendorf, F.W.; Holt, J.S.; Lodge, D.M.; Molofsky, J.; With, K.A.; Baughman, S.; Cabin, R.J.; Cohen, J.E.; Ellstrand, N.C.; et al. The population biology of invasive species. Annu. Rev. Ecol. Syst. 2001, 32, 305–332. [Google Scholar] [CrossRef]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Crotti, S.; Piccoli, M.; Rizzolio, F.; Giordano, A.; Nitti, D.; Agostini, M. Extracellular matrix and colorectal cancer: How surrounding microenvironment affects cancer cell behavior? J. Cell. Physiol. 2017, 232, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Quaranta, V. Tales from the crypt[ic] sites of the extracellular matrix. Trends Cell Biol. 2003, 13, 366–375. [Google Scholar] [CrossRef]
- Li, Z.; Lee, H.; Zhu, C. Molecular mechanisms of mechanotransduction in integrin-mediated cell-matrix adhesion. Exp. Cell Res. 2016, 349, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.T.; Thakar, D.; Weaver, V.M. Force-dependent breaching of the basement membrane. Matrix Biol. 2017, 57–58, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Mekhdjian, A.H.; Kai, F.; Rubashkin, M.G.; Prahl, L.S.; Przybyla, L.M.; McGregor, A.L.; Bell, E.S.; Matthew, B.; DuFort, C.C.; Ou, G.; et al. Integrin-mediated traction force enhances paxillin molecular associations and adhesion dynamics that increase the invasiveness of tumor cells into a three-dimensional extracellular matrix. Mol. Biol. Cell 2017, 28, 1467–1488. [Google Scholar] [CrossRef] [PubMed]
- Walter, C.; Crawford, L.; Lai, M.; Toonen, J.A.; Pan, Y.; Sakiyama-Elbert, S.; Gutmann, D.H.; Pathak, A. Increased tissue stiffness in tumors from mice with Neurofibromatosis-1 optic glioma. Biophys. J. 2017, 112, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Marangon, I.; Silva, A.A.K.; Guilbert, T.; Kolosnjaj-Tabi, J.; Marchiol, C.; Natkhunarajah, S.; Chamming’s, F.; Ménard-Moyon, C.; Bianco, A.; Gennisson, J.-L.; et al. Tumor stiffening, a key determinant of tumor progression, is reversed by nanomaterial-induced photothermal therapy. Theranostics 2017, 7, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Relation, T.; Dominici, M.; Horwitz, E.M. Concise review: An (Im)Penetrable shield: How the tumor microenvironment protects cancer stem cells. Stem Cells 2017, 35, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.G.; Nedel, F.; Alves, A.M.; Nör, J.E.; Tarquinio, S.B.C. Tumor angiogenesis and lymphangiogenesis: Tumor/endothelial crosstalk and cellular/microenvironmental signaling mechanisms. Life Sci. 2013, 92, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Tosetti, F.; Benelli, R.; Noonan, D.M. Tumor inflammatory angiogenesis and its chemoprevention. Cancer Res. 2005, 65, 10637–10641. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Wang, C.-Y.; Werb, Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol. 2015, 44–46, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Ishii, G.; Ochiai, A.; Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 2016, 99, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Heindl, A.; Nawaz, S.; Yuan, Y. Mapping spatial heterogeneity in the tumor microenvironment: A new era for digital pathology. Lab. Investig. 2015, 95, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Tabassum, D.P.; Altrock, P.M.; Almendro, V.; Michor, F.; Polyak, K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014, 514, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Tomaso, E.D.; Capen, D.; Haskell, A.; Hart, J.; Logie, J.J.; Jain, R.K.; McDonald, D.M.; Jones, R.; Munn, L.L. Mosaic tumor vessels: Cellular basis and ultrastructure of focal regions lacking endothelial cell markers. Cancer Res. 2005, 65, 5740–5749. [Google Scholar] [CrossRef] [PubMed]
- Clemente, M.; Pérez-Alenza, M.D.; Illera, J.C.; Peña, L. Histological, immunohistological, and ultrastructural description of vasculogenic mimicry in canine mammary cancer. Vet. Pathol. 2010, 47, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Cui, J.; Barnes, L.; Cheresh, D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J. Cell Biol. 2004, 167, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Selby, P.J.; Thomas, J.M.; Monaghan, P.; Sloane, J.; Peckham, M.J. Human tumour xenografts established and serially transplanted in mice immunologically deprived by thymectomy, cytosine arabinoside and whole-body irradiation. Br. J. Cancer 1980, 41, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, P.; Raghavan, D.; Neville, A.M. Ultrastructural studies of xenografted human germ cell tumors. Cancer 1982, 49, 683–697. [Google Scholar] [CrossRef]
- Russell, P.J.; Raghavan, D.; Gregory, P.; Philips, J.; Wills, E.J.; Jelbart, M.; Wass, J.; Zbroja, R.A.; Vincent, P.C. Bladder cancer xenografts: A model of tumor cell heterogeneity. Cancer Res. 1986, 46, 2035–2040. [Google Scholar] [PubMed]
- Steinberg, F.; Konerding, M.A.; Streffer, C. The vascular architecture of human xenotransplanted tumors: Histological, morphometrical, and ultrastructural studies. J. Cancer Res. Clin. Oncol. 1990, 116, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Lièvre, C.S.L.; Douarin, N.M.L. Mesenchymal derivatives of the neural crest: Analysis of chimaeric quail and chick embryos. Development 1975, 34, 125–154. [Google Scholar]
- Dauer, D.J.; Ferraro, B.; Song, L.; Yu, B.; Mora, L.; Buettner, R.; Enkemann, S.; Jove, R.; Haura, E.B. Stat3 regulates genes common to both wound healing and cancer. Oncogene 2005, 24, 3397–3408. [Google Scholar] [CrossRef] [PubMed]
- Kulesa, P.M.; Morrison, J.A.; Bailey, C.M. The neural crest and cancer: A developmental spin on melanoma. Cells Tissues Organs 2013, 198, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.J.; Lessey, B.A. Embryo implantation and tumor metastasis: Common pathways of invasion and angiogenesis. Semin. Reprod. Endocrinol. 1999, 17, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tan, C.; Xiao, F.; Zou, L.; Wang, L.; Wei, Y.; Yang, H.; Zhang, W. The “inherent vice” in the anti-angiogenic theory may cause the highly metastatic cancer to spread more aggressively. Sci. Rep. 2017, 7, 2365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhong, B.; Chen, M.; Yang, L.; Yang, G.; Li, Y.; Wang, H.; Wang, G.; Li, W.; Cui, J.; et al. Epigenetic reprogramming reverses the malignant epigenotype of the MMP/TIMP axis genes in tumor cells. Int. J. Cancer 2014, 134, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Aktipis, C.A.; Boddy, A.M.; Gatenby, R.A.; Brown, J.S.; Maley, C.C. Life history tradeoffs in cancer evolution. Nat. Rev. Cancer 2013, 13, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef] [PubMed]
- Saltel, F.; Daubon, T.; Juin, A.; Ganuza, I.E.; Veillat, V.; Génot, E. Invadosomes: Intriguing structures with promise. Eur. J. Cell Biol. 2011, 90, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A. Invadopodia: Specialized cell structures for cancer invasion. Clin. Exp. Metastasis 2006, 23, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Hegerfeldt, Y.; Tusch, M.; Bröcker, E.-B.; Friedl, P. Collective cell movement in primary melanoma explants: Plasticity of cell-cell interaction, β1-integrin function, and migration strategies. Cancer Res. 2002, 62, 2125–2130. [Google Scholar] [PubMed]
- Chapman, A.; Fernandez del Ama, L.; Ferguson, J.; Kamarashev, J.; Wellbrock, C.; Hurlstone, A. Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep. 2014, 8, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Lintz, M.; Muñoz, A.; Reinhart-King, C.A. The mechanics of single cell and collective migration of tumor cells. J. Biomech. Eng. 2017, 139, 021005. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, P.; Peng, J.; Wang, X.; Zhu, Y.; Shen, N. Meta-analysis reveals the prognostic value of circulating tumour cells detected in the peripheral blood in patients with non-metastatic colorectal cancer. Sci. Rep. 2017, 7, 905. [Google Scholar] [CrossRef] [PubMed]
- Pernot, S.; Badoual, C.; Terme, M.; Castan, F.; Cazes, A.; Bouche, O.; Bennouna, J.; Francois, E.; Ghiringhelli, F.; Fouchardiere, C.D.L.; et al. Dynamic evaluation of circulating tumour cells in patients with advanced gastric and oesogastric junction adenocarcinoma: Prognostic value and early assessment of therapeutic effects. Eur. J. Cancer 2017, 79, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Nauta, A.J.; Fibbe, W.E. Immunomodulatory properties of mesenchymal stromal cells. Blood 2007, 110, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Villanova, L.; De Maria, R. Cancer stem cells: At the forefront of personalized medicine and immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Koopman, C.D.; Werb, Z. Circulating tumor cells. Science 2013, 341, 1186–1188. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi, A.A.; Sahebkar, A.; Gholamin, M. Isolation, identification, and characterization of cancer stem cells: A review. J. Cell. Physiol. 2017, 232, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.-C.; Pierga, J.-Y.; Vincent-Salomon, A.; Poupon, M.-F. A “class action” against the microenvironment: Do cancer cells cooperate in metastasis? Cancer Metastasis Rev. 2008, 27, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; He, S.; Zhang, L.; Snaar-Jagalska, B.E.; ten Dijke, P. Transforming growth factor-β signalling controls human breast cancer metastasis in a zebrafish xenograft model. Breast Cancer Res. 2013, 15, R106. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-H.; Chen, Y.-D.; Huang, S.-F.; Wang, H.-M.; Wu, M.-H. The Effect of Primary Cancer Cell Culture Models on the Results of Drug Chemosensitivity Assays: The Application of Perfusion Microbioreactor System as Cell Culture Vessel. Available online: https://www.hindawi.com/journals/bmri/2015/470283/ (accessed on 21 August 2017).
- Chitcholtan, K.; Asselin, E.; Parent, S.; Sykes, P.H.; Evans, J.J. Differences in growth properties of endometrial cancer in three dimensional (3D) culture and 2D cell monolayer. Exp. Cell Res. 2013, 319, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Aljitawi, O.S.; Li, D.; Xiao, Y.; Zhang, D.; Ramachandran, K.; Stehno-Bittel, L.; Van Veldhuizen, P.; Lin, T.L.; Kambhampati, S.; Garimella, R. A novel three-dimensional stromal-based model for in vitro chemotherapy sensitivity testing of leukemia cells. Leuk. Lymphoma 2014, 55, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. The relevance of using 3D cell cultures, in addition to 2D monolayer cultures, when evaluating breast cancer drug sensitivity and resistance. Oncotarget 2016, 7, 45745–45756. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Ramaiahgari, S.C.; Braver, M.W.; Herpers, B.; Terpstra, V.; Commandeur, J.N.M.; van de Water, B.; Price, L.S. A 3D In Vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch. Toxicol. 2014, 88, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Brugge, J.S. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 2005, 5, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Daniel, V.C.; Marchionni, L.; Hierman, J.S.; Rhodes, J.T.; Devereux, W.L.; Rudin, C.M.; Yung, R.; Parmigiani, G.; Dorsch, M.; Peacock, C.D.; et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture In vitro. Cancer Res. 2009, 69, 3364–3373. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Ince, T.A. Of mice and women: A comparative tissue biology perspective of breast stem cells and differentiation. J. Mammary Gland Biol. Neoplasia 2015, 20, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Kuracha, M.R.; Thomas, P.; Loggie, B.W.; Govindarajan, V. Patient-derived xenograft mouse models of pseudomyxoma peritonei recapitulate the human inflammatory tumor microenvironment. Cancer Med. 2016, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Lorsch, J.R.; Collins, F.S.; Lippincott-Schwartz, J. Fixing problems with cell lines. Science 2014, 346, 1452–1453. [Google Scholar] [CrossRef] [PubMed]
- Freedman, L.P.; Gibson, M.C.; Ethier, S.P.; Soule, H.R.; Neve, R.M.; Reid, Y.A. Reproducibility: Changing the policies and culture of cell line authentication. Nat. Methods 2015, 12, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Freedman, L.P.; Cockburn, I.M.; Simcoe, T.S. The economics of reproducibility in preclinical research. PLoS Biol. 2015, 13, e1002165. [Google Scholar] [CrossRef] [PubMed]
- Wilding, J.L.; Bodmer, W.F. Cancer cell lines for drug discovery and development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Tveit, K.M.; Pihl, A. Do cell lines in vitro reflect the properties of the tumours of origin? A study of lines derived from human melanoma xenografts. Br. J. Cancer 1981, 44, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, M.; Satake, H.; Uchida, J.; Shimamoto, Y.; Kato, T.; Takechi, T.; Okabe, H.; Fujioka, A.; Nakano, K.; Ohshimo, H.; et al. Preclinical antitumor efficacy of S-1: A new oral formulation of 5-fluorouracil on human tumor xenografts. Int. J. Oncol. 1998, 13, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Gaudenzi, G.; Albertelli, M.; Dicitore, A.; Würth, R.; Gatto, F.; Barbieri, F.; Cotelli, F.; Florio, T.; Ferone, D.; Persani, L.; et al. Patient-derived xenograft in zebrafish embryos: A new platform for translational research in neuroendocrine tumors. Endocrine 2017, 57, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Corthay, A. Does the immune system naturally protect against cancer? Front. Immunol. 2014, 5, 197. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, C.; Song, Y.; Wang, Z.; Wang, Y.; Luo, F.; Xu, Y.; Zhao, Y.; Wu, Z.; Xu, Y. Mechanism of immune evasion in breast cancer. OncoTargets Ther. 2017, 10, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Childs, R.W.; Carlsten, M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: The force awakens. Nat. Rev. Drug Discov. 2015, 14, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.J.; Shklovskaya, E.; Hersey, P. Epigenetic modulation in cancer immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; O’Neill, W.; Pan, Q. Immunotherapy for head and neck cancer: The future of treatment? Expert Opin. Biol. Ther. 2017, 17, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Zito, M.; Ascierto, P.A.; Rossi, G.; Staibano, S.; Montella, M.; Russo, D.; Alfano, R.; Morabito, A.; Botti, G.; Franco, R. Are tumor-infiltrating lymphocytes protagonists or background actors in patient selection for cancer immunotherapy? Expert Opin. Biol. Ther. 2017, 17, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.C.; Langenau, D.M. Allograft cancer cell transplantation in zebrafish. Adv. Exp. Med. Biol. 2016, 916, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shimada, Y.; Hirota, T.; Ariyoshi, M.; Kuroyanagi, J.; Nishimura, Y.; Tanaka, T. Novel immunologic tolerance of human cancer cell xenotransplants in zebrafish. Transl. Res. 2016, 170, 89–98. [Google Scholar] [CrossRef] [PubMed]
- White, R.M.; Sessa, A.; Burke, C.; Bowman, T.; LeBlanc, J.; Ceol, C.; Bourque, C.; Dovey, M.; Goessling, W.; Burns, C.E.; et al. Transparent adult zebrafish as a tool for In Vivo transplantation analysis. Cell Stem Cell 2008, 2, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, S.; Ratnakumar, K.; Langdon, E.M.; Kansler, E.R.; Kim, I.S.; Campbell, N.R.; Perry, E.B.; McMahon, A.J.; Kaufman, C.K.; van Rooijen, E.; et al. A Quantitative system for studying metastasis using transparent zebrafish. Cancer Res. 2015, 75, 4272–4282. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.S.; de Peer, Y.V.; Braasch, I.; Meyer, A. Comparative genomics provides evidence for an ancient genome duplication event in fish. Philos. Trans. R. Soc. Lond. B 2001, 356, 1661–1679. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, S.K.; Neuhauss, S.F. Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genom. 2014, 289, 1045–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.S.; Braasch, I.; Frickey, T.; Meyer, A.; Van de Peer, Y. Genome duplication, a trait shared by 22,000 species of ray-finned fish. Genome Res. 2003, 13, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.S.; Raes, J. Duplication and divergence: The evolution of new genes and old ideas. Annu. Rev. Genet. 2004, 38, 615–643. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.D.; Gentleman, E. The role of material structure and mechanical properties in cell-matrix interactions. J. Mater. Chem. B 2014, 2, 2345–2356. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, R.A.; Keow, J.Y.; Harris, N.D.; Haché, C.A.; Li, D.H.; Crawford, B.D. The zebrafish embryo: A powerful model system for investigating matrix remodeling. Zebrafish 2009, 6, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Klupp, F.; Neumann, L.; Kahlert, C.; Diers, J.; Halama, N.; Franz, C.; Schmidt, T.; Koch, M.; Weitz, J.; Schneider, M.; et al. Serum MMP7, MMP10 and MMP12 level as negative prognostic markers in colon cancer patients. BMC Cancer 2016, 16, 494. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Li, Z.; Lin, L.; Lei, Y.; Hongyuan, Y.; Hongwei, J.; Yang, L.; Chuize, K. MMP1, 2, 3, 7, and 9 gene polymorphisms and urinary cancer risk: A meta-analysis. Genet. Test. Mol. Biomark. 2015, 19, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Liu, C. Pathological and prognostic significance of matrix metalloproteinase-2 expression in ovarian cancer: A meta-analysis. Clin. Exp. Med. 2015, 16, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, D.Y.; Zhou, W.L.; Wang, M.W.; Xia, W.M.; Tang, Q. Prognostic value of matrix metalloprotease-1/protease-activated receptor-1 axis in patients with prostate cancer. Med. Oncol. 2014, 31, 968. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, S.T.; Sizemore, G.M.; Booth, C.N.; Thompson, C.L.; Silverman, P.; Bebek, G.; Abdul-Karim, F.W.; Avril, S.; Keri, R.A. Hypomethylation of the MMP7 promoter and increased expression of MMP7 distinguishes the basal-like breast cancer subtype from other triple-negative tumors. Breast Cancer Res. Treat. 2014, 146, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Puzovic, V.; Brcic, I.; Ranogajec, I.; Jakic-Razumovic, J. Prognostic values of ETS-1, MMP-2 and MMP-9 expression and co-expression in breast cancer patients. Neoplasma 2014, 61, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Koskensalo, S.; Mrena, J.; Wiksten, J.-P.; Nordling, S.; Kokkola, A.; Hagström, J.; Haglund, C. MMP-7 overexpression is an independent prognostic marker in gastric cancer. Tumor Biol. 2010, 31, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Polistena, A.; Cucina, A.; Dinicola, S.; Stene, C.; Cavallaro, G.; Ciardi, A.; Orlando, G.; Arena, R.; D’Ermo, G.; Cavallaro, A.; et al. MMP7 expression in colorectal tumours of different stages. In Vivo 2014, 28, 105–110. [Google Scholar] [PubMed]
- Apte, S.S.; Parks, W.C. Metalloproteinases: A parade of functions in matrix biology and an outlook for the future. Matrix Biol. 2015, 44–46, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015, 44–46, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Bekes, E.M.; Deryugina, E.I.; Kupriyanova, T.A.; Zajac, E.; Botkjaer, K.A.; Andreasen, P.A.; Quigley, J.P. Activation of pro-uPA is critical for initial escape from the primary tumor and hematogenous dissemination of human carcinoma cells. Neoplasia 2011, 13, 806–821. [Google Scholar] [CrossRef] [PubMed]
- Sodek, K.L.; Ringuette, M.J.; Brown, T.J. MT1-MMP is the critical determinant of matrix degradation and invasion by ovarian cancer cells. Br. J. Cancer 2007, 97, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rossé, C.; Chavrier, P. Cellular and molecular mechanisms of MT1-MMP-dependent cancer cell invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef] [PubMed]
- Leigh, N.R.; Schupp, M.-O.; Li, K.; Padmanabhan, V.; Gastonguay, A.; Wang, L.; Chun, C.Z.; Wilkinson, G.A.; Ramchandran, R. Mmp17b is essential for proper neural crest cell migration In Vivo. PLoS ONE 2013, 8, e76484. [Google Scholar] [CrossRef] [PubMed]
- Crawford, B.D.; Po, M.D.; Saranyan, P.V.; Forsberg, D.; Schulz, R.; Pilgrim, D.B. Mmp25β facilitates elongation of sensory neurons during zebrafish development. Genesis 2014, 52, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Keow, J.Y.; Crawford, B.D. Investigating matrix metalloproteinase regulation in its biological context; Detecting MMP activity In Vivo. In Matrix Metalloproteinases; Oshiro, N., Miyagi, E., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2012; pp. 151–169. ISBN 978-1-62100-789-0. [Google Scholar]
- Nguyen, T.T.-T.N.; Shynlova, O.; Lye, S.J. Matrix metalloproteinase expression in the rat myometrium during pregnancy, term labor, and postpartum. Biol. Reprod. 2016, 95, 24. [Google Scholar] [CrossRef] [PubMed]
- Hulboy, D.L.; Rudolph, L.A.; Matrisian, L.M. Matrix metalloproteinases as mediators of reproductive function. MHR Basic Sci. Reprod. Med. 1997, 3, 27–45. [Google Scholar] [CrossRef]
- Chiang, K.-C.; Hsu, S.-Y.; Lin, S.-J.; Yeh, C.-N.; Pang, J.-H.; Wang, S.-Y.; Hsu, J.-T.; Yeh, T.-S.; Chen, L.-W.; Kuo, S.-F.; et al. PTEN insufficiency increases breast cancer cell metastasis in vitro and In Vivo in a xenograft zebrafish model. Anticancer Res. 2016, 36, 3997–4005. [Google Scholar] [PubMed]
- Cock-Rada, A.M.; Medjkane, S.; Janski, N.; Yousfi, N.; Perichon, M.; Chaussepied, M.; Chluba, J.; Langsley, G.; Weitzman, J.B. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res 2012, 72, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Wu, Y.-N.; Wang, S.-L.; Lin, Q.-H.; He, M.-F.; Liu, Q.-L.; Wang, J.-H. Docosahexaenoic acid modulates invasion and metastasis of human ovarian cancer via multiple molecular pathways. Int. J. Gynecol. Cancer 2016, 26, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.E.; Junttila, M.R.; de Sauvage, F.J. Translational value of mouse models in oncology drug development. Nat. Med. 2015, 21, 431–439. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| MMP | Zebrafish Representation | MMP | Zebrafish Representation |
|---|---|---|---|
| 2 | * | 1 | - |
| 9 | * | 8 | - |
| 14 | ** | 13 | ** |
| 15 | * | 19 | * |
| 16 | ** | 7 | - |
| 17 | ** | 26 | - |
| 24 | * | 20 | ** |
| 25 | ** | 21 | * |
| 3 | - | 23 | ** |
| 10 | - | 27 | - |
| 11 | ** | 28 | * |
| 12 | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wyatt, R.A.; Trieu, N.P.V.; Crawford, B.D. Zebrafish Xenograft: An Evolutionary Experiment in Tumour Biology. Genes 2017, 8, 220. https://doi.org/10.3390/genes8090220
Wyatt RA, Trieu NPV, Crawford BD. Zebrafish Xenograft: An Evolutionary Experiment in Tumour Biology. Genes. 2017; 8(9):220. https://doi.org/10.3390/genes8090220
Chicago/Turabian StyleWyatt, Rachael A., Nhu P. V. Trieu, and Bryan D. Crawford. 2017. "Zebrafish Xenograft: An Evolutionary Experiment in Tumour Biology" Genes 8, no. 9: 220. https://doi.org/10.3390/genes8090220
APA StyleWyatt, R. A., Trieu, N. P. V., & Crawford, B. D. (2017). Zebrafish Xenograft: An Evolutionary Experiment in Tumour Biology. Genes, 8(9), 220. https://doi.org/10.3390/genes8090220