Splicing Analysis of Exonic OCRL Mutations Causing Lowe Syndrome or Dent-2 Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioinformatics Predictions
2.2. Amplification of OCRL Genomic Fragments
2.3. Generation of Minigene Constructs
2.4. Site-Directed Mutagenesis
2.5. Cell Culture, Transient Transfection and RT-PCR Assay
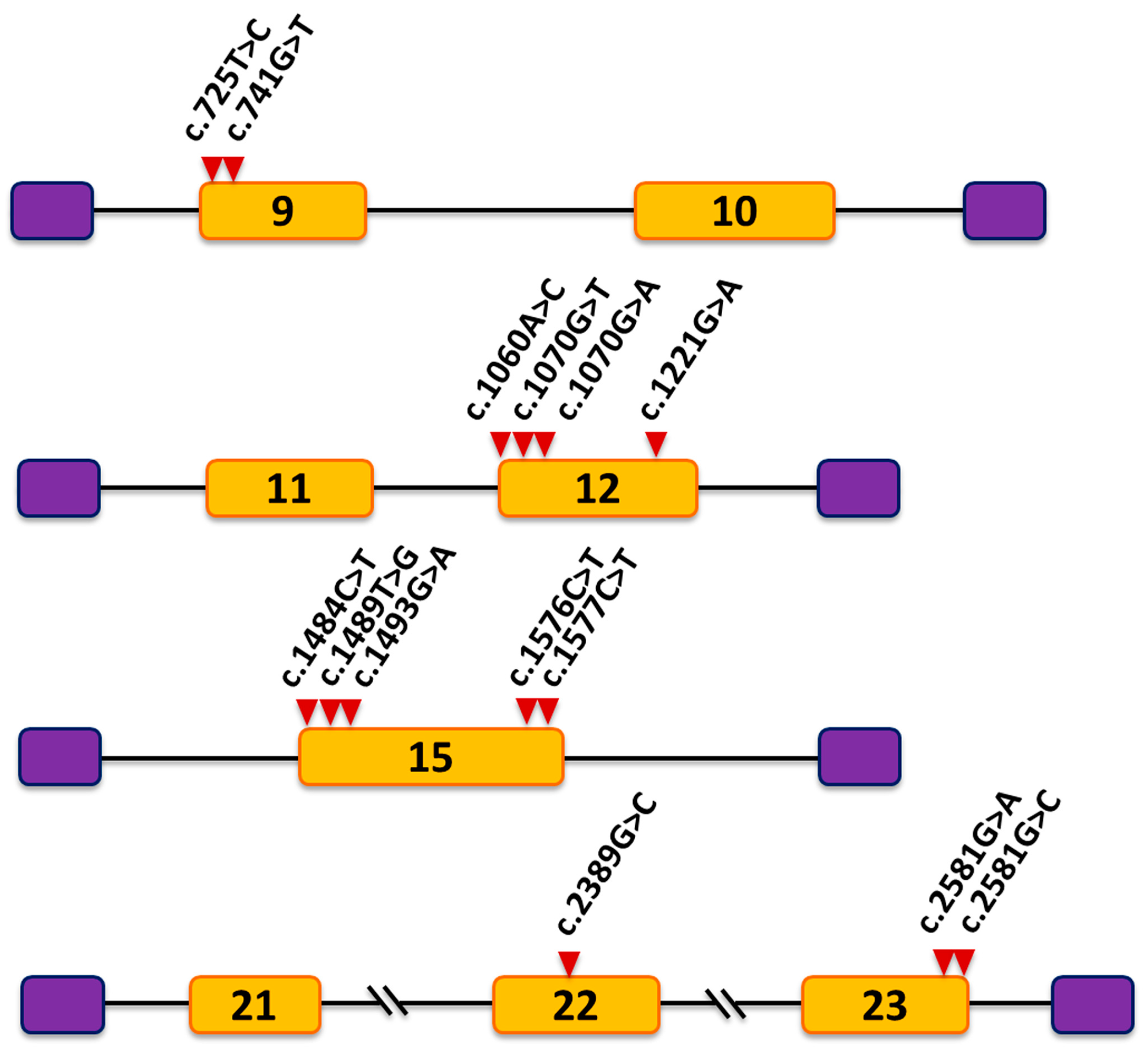
3. Results
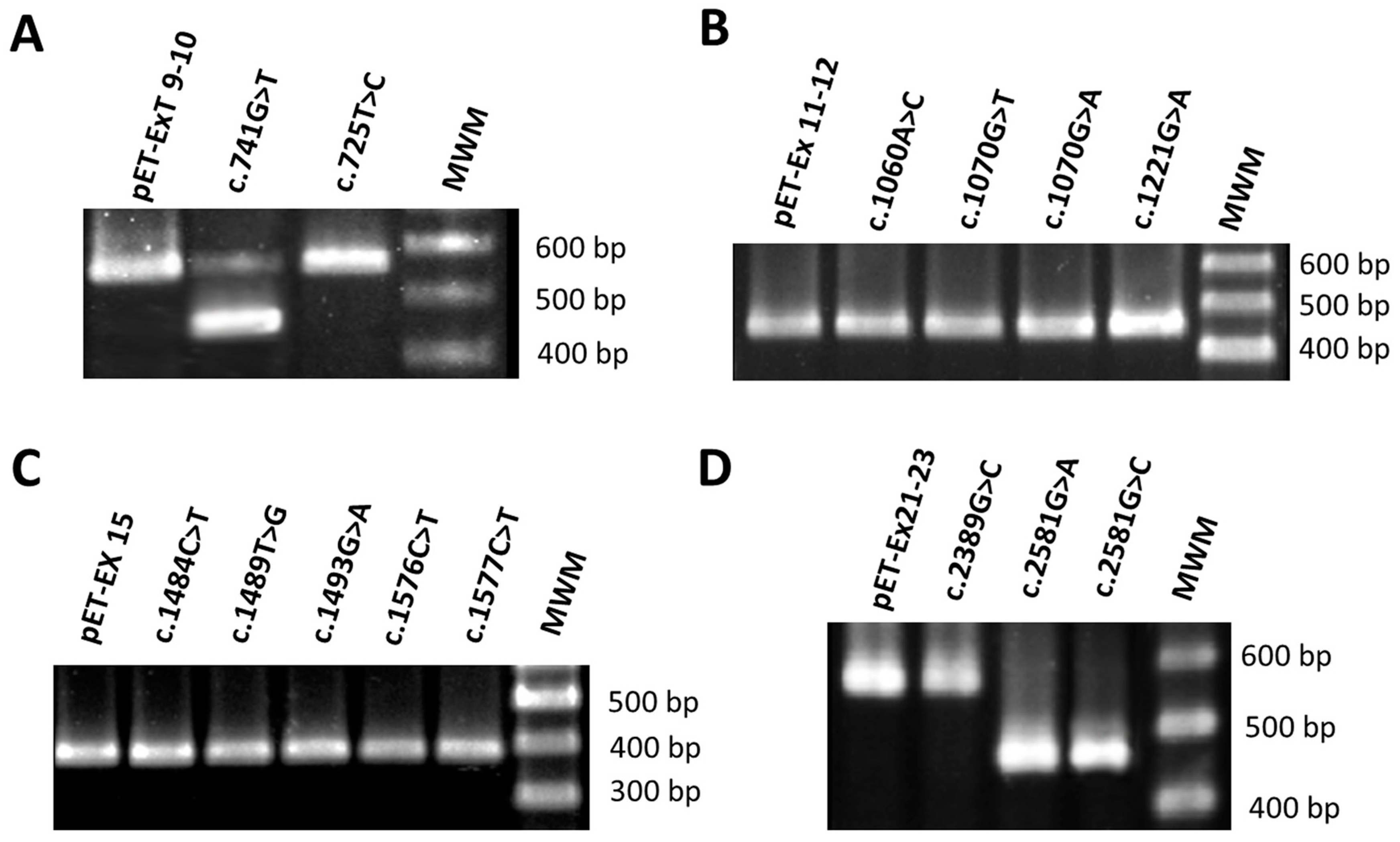
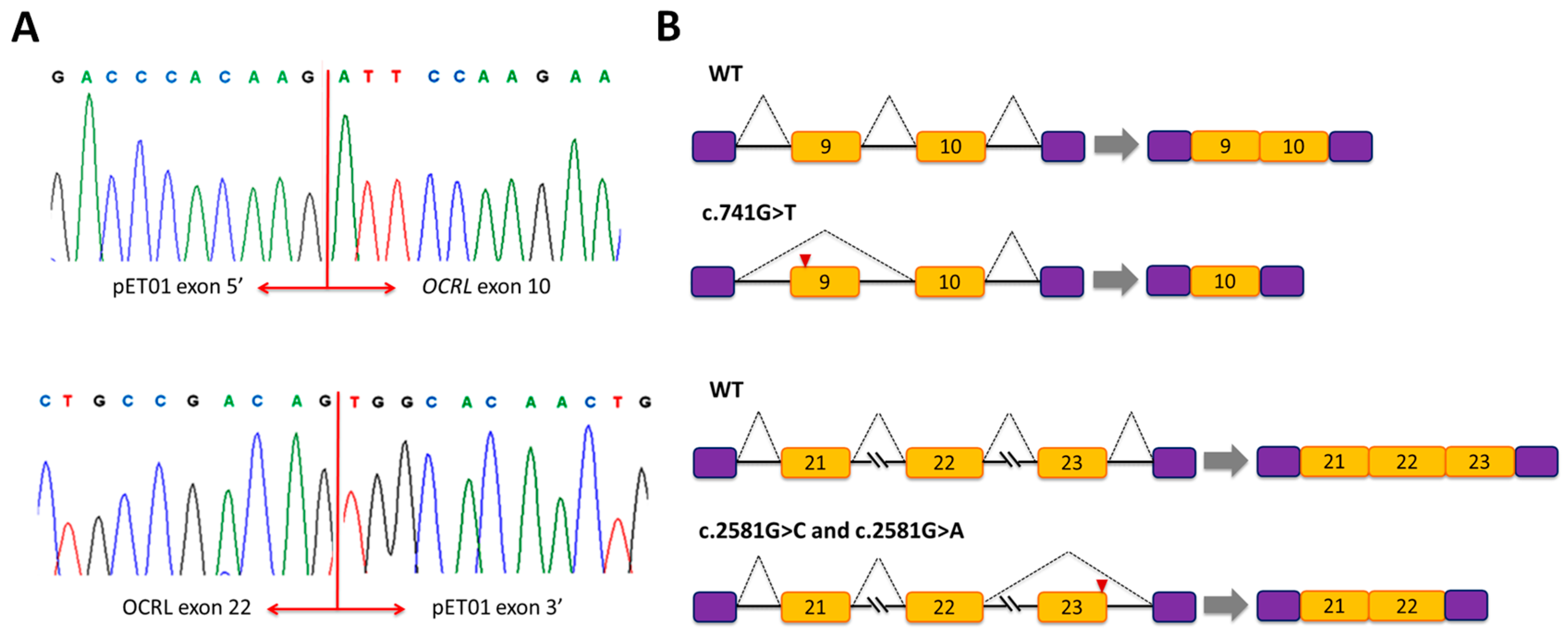
3.1. Mutation c.741G>T Prevents Incorporation of Exon 9 into the Mature mRNA
3.2. Mutations c.2581G>A and c.2581G>C Result in Skipping of Exon 23
3.3. Mutations in Exons 12, 15 and 22 Did Not Alter Pre-mRNA Splicing
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affects splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Sterne-Weiler, T.; Howard, J.; Mort, M.; Cooper, D.N.; Sanford, J.R. Loss of exon identity is a common mechanism of human inherited disease. Genome Res. 2011, 21, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Claverie-Martin, F.; Gonzalez-Paredes, F.J.; Ramos-Trujillo, E. Splicing defects caused by exonic mutations in PKD1 as a new mechanism of pathogenesis in autosomal dominant polycystic kidney disease. RNA Biol. 2015, 12, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Claverie-Martin, F.; Ramos-Trujillo, E.; García-Nieto, V. Dent’s disease: Clinical features and molecular basis. Pediatr. Nephrol. 2011, 26, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Bökenkamp, A.; Ludwig, M. The oculocerebrorenal syndrome of Lowe: An update. Pediatr. Nephrol. 2016, 31, 2201–2212. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, M.A.; Staiano, L.; Emma, F.; Devuyst, O. The 5-phosphatase OCRL in Lowe syndrome and Dent disease 2. Nat. Rev. Nephrol. 2017, 13, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Bökenkamp, A.; Böckenhauer, D.; Cheong, H.I.; Hoppe, B.; Tasic, V.; Unwin, R.; Ludwig, M. Dent-2 disease: A mild variant of Lowe syndrome. J. Pediatr. 2009, 155, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Shrimpton, A.E.; Hoopes, R.R., Jr.; Knohl, S.J.; Hueber, P.; Reed, A.A.; Christie, P.T.; Igarashi, T.; Lee, P.; Lehman, A.; White, C.; et al. OCRL1 mutations in Dent2 patients suggest a mechanism for phenotypic variability. Nephron Physiol. 2009, 112, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Bockenhauer, D.; Bokenkamp, A.; van’t Hoff, W.; Levtchenko, E.; Kist-van Holthe, J.E.; Tasic, V.; Ludwig, M. Renal phenotype in Lowe Syndrome: A selective proximal tubular dysfunction. Clin. J. Am. Soc. Nephrol. 2008, 3, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Attree, O.; Olivos, I.M.; Okabe, I.; Bailey, L.C.; Nelson, D.L.; Lewis, R.A.; McInnes, R.R.; Nussbaum, R.L. The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 1992, 358, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Hoopes, R.R., Jr.; Shrimpton, A.E.; Knohl, S.J.; Hueber, P.; Hoppe, B.; Matyus, J.; Simckes, A.; Tasic, V.; Toenshoff, B.; Suchy, S.F.; et al. Dent Disease with mutations in OCRL1. Am. J. Hum. Genet. 2005, 76, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jefferson, A.B.; Auethavekiat, V.; Majerus, P.W. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc. Natl. Acad. Sci. USA 1995, 92, 4853–4856. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, R.L.; Orrison, B.M.; Jänne, P.A.; Charnas, L.; Chinault, A.C. Physical mapping and genomic structure of the Lowe syndrome gene OCRL1. Hum. Genet. 1997, 99, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, R.; Noakes, C.J.; McKenzie, E.; Kox, C.; Lowe, M. Differential clathrin binding and subcellular localization of OCRL1 splice isoforms. J. Biol. Chem. 2009, 284, 9965–9973. [Google Scholar] [CrossRef] [PubMed]
- Mehta, Z.B.; Pietka, G.; Lowe, M. The cellular and physiological functions of the Lowe syndrome protein OCRL1. Traffic 2014, 15, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Ball, E.V.; Stenson, P.D.; Phillips, A.D.; Evans, K.; Heywood, S.; Hayden, M.J.; Azevedo, L.; Mort, M.E.; Hussain, M. The Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 9 November 2017).
- Hichri, H.; Rendu, J.; Monnier, N.; Coutton, C.; Dorseuil, O.; Poussou, R.V.; Baujat, G.; Blanchard, A.; Nobili, F.; Ranchin, B.; et al. From Lowe syndrome to Dent disease: Correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum. Mutat. 2011, 32, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaniew, M.; Bökenkamp, A.; Kołbuc, M.; La Scola, C.; Baronio, F.; Niemirska, A.; Szczepańska, M.; Bürger, J.; La Manna, A.; Miklaszewska, M.; et al. Long-term renal outcome in children with OCRL mutations: Retrospective analysis of a large international cohort. Nephrol. Dial. Transplant. 2016, 5, gfw350. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.T. Sequence variant nomenclature. Available online: http://varnomen.hgvs.org/ (accessed on 9 November 2017).
- Recker, F.; Zaniew, M.; Böckenhauer, D.; Miglietti, N.; Bökenkamp, A.; Moczulska, A.; Rogowska-Kalisz, A.; Laube, G.; Said-Conti, V.; Kasap-Demir, B.; et al. Characterization of 28 novel patients expands the mutational and phenotypic spectrum of Lowe syndrome. Pediatr. Nephrol. 2015, 30, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Claverie-Martin, F. Identification of a New Synonymous OCRL Variant in a Patient with Dent Disease; Unidad de Investigación, Hospital Nuestra Señora de Candelaria: Santa Cruz de Tenerife, Spain, 2015. [Google Scholar]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucl. Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef] [PubMed]
- Fairbrother, W.G.; Yeh, R.F.; Sharp, P.A.; Burge, C.B. Predictive identification of exonic splicing enhancers in human genes. Science 2002, 297, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Rolish, M.E.; Yeo, G.; Tung, V.; Mawson, M.; Burge, C.B. Systematic identification and analysis of exonic splicing silencers. Cell 2004, 119, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Mort, M.; Sterne-Weiler, T.; Li, B.; Ball, E.V.; Cooper, D.N.; Radivojac, P.; Sanford, J.R.; Mooney, S.D. MutPred Splice: Machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol. 2014, 15, R19. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Gene runner. Available online: http://generunner.net (accessed on 9 November 2017).
- QuikChange Primer Design. Available online: http://www.genomics.agilent.com/primerDesignProgram.jsp (accessed on 9 November 2017).
- Basic Local Alignment Search Tool (BLAST). Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 9 November 2017).
- Wu, Y.; Zhang, Y.; Zhang, J. Distribution of exonic splicing enhancer elements in human genes. Genomics 2005, 86, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Paredes, F.J.; Ramos-Trujillo, E.; Claverie-Martin, F. Defective pre-mRNA splicing in PKD1 due to presumed missense and synonymous mutations causing autosomal dominant polycystic disease. Gene 2014, 546, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Addis, M.; Loi, M.; Lepiani, C.; Cau, M.; Melis, M.A. OCRL mutation analysis in Italian patients with Lowe syndrome. Hum. Mutat. 2004, 23, 524–525. [Google Scholar] [CrossRef] [PubMed]
- Monnier, N.; Satre, V.; Lerouge, E.; Berthoin, F.; Lunardi, J. OCRL1 mutation analysis in French Lowe syndrome patients: Implications for molecular diagnosis strategy and genetic counseling. Hum. Mutat. 2000, 16, 157–165. [Google Scholar] [CrossRef]
- Tasic, V.; Lozanovski, V.J.; Korneti, P.; Ristoska-Bojkovska, N.; Sabolic-Avramovska, V.; Gucev, Z.; Ludwig, M. Clinical and laboratory features of Macedonian children with OCRL mutations. Pediatr. Nephrol. 2011, 26, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.F.; Boltshauser, E.; Neuhaus, T.J.; Rauscher, C.; Martin, E. MRI and proton spectroscopy in Lowe syndrome. Neuropediatrics 2001, 32, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Hasui, M.; Hata, A.; Hata, D.; Nozu, K. Focal segmental glomerulosclerosis in a boy with Dent-2 disease. Pediatr. Nephrol. 2010, 25, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Röschinger, W.; Muntau, A.C.; Rudolph, G.; Roscher, A.A.; Kammerer, S. Carrier assessment in families with lowe oculocerebrorenal syndrome: Novel mutations in the OCRL1 gene and correlation of direct DNA diagnosis with ocular examination. Mol. Genet. Metab. 2000, 69, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucl. Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Lindeboom, R.G.H.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Pagenstecher, C.; Wehner, M.; Friedl, W.; Rahner, N.; Aretz, S.; Friedrichs, N.; Sengteller, M.; Henn, W.; Buettner, R.; Propping, P.; et al. Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum. Genet. 2006, 119, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Ferraris, L.; Filloux, M.E.; Raphael, B.J.; Fairbrother, W.G. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc. Natl. Acad. Sci. USA 2011, 108, 11093–11098. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Buratti, E. RNA splicing in human disease and in the clinic. Clin. Sci. 2017, 131, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Lastella, P.; Surdo, N.C.; Resta, N.; Guanti, G.; Stella, A. In silico and in vivo splicing analysis of MLH1 and MSH2 missense mutations shows exon- and tissue-specific effects. BMC Genom. 2006, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Skryabin, B.V.; Müller, T.; Gazinski, A.; Schröter, J.; Gassner, B.; Nikolaev, V.O.; Bünemann, M.; Kuhn, M. Alternative splicing of the guanylyl cyclase-A receptor modulates atrial natriuretic peptide signaling. J. Biol. Chem. 2008, 283, 28313–28320. [Google Scholar] [CrossRef] [PubMed]
- Houdayer, C.; Dehainault, C.; Mattler, C.; Michaux, D.; Caux-Moncoutier, V.; Pagès-Berhouet, S.; d’Enghien, C.D.; Laugé, A.; Castera, L.; Gauthier-Villars, M.; et al. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum. Mutat. 2008, 29, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Gaildrat, P.; Killian, A.; Martins, A.; Tournier, I.; Frébourg, T.; Tosi, M. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol. Biol. 2010, 653, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Tournier, I.; Vezain, M.; Martins, A.; Charbonnier, F.; Baert-Desurmont, S.; Olschwang, S.; Wang, Q.; Buisine, M.P.; Soret, J.; Tazi, J.; et al. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum. Mutat. 2008, 29, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Krieger, S.; Vezain, M.; Rousselin, A.; Tournier, I.; Martins, A.; Berthet, P.; Chevrier, A.; Dugast, C.; Layet, V.; et al. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J. Med. Genet. 2008, 45, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Van der Klift, H.M.; Jansen, A.M.; van der Steenstraten, N.; Bik, E.C.; Tops, C.M.; Devilee, P.; Wijnen, J.T. Splicing analysis for exonic and intronic mismatch repair gene variants associated with Lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Mol. Genet. Genom. Med. 2015, 3, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Théry, J.C.; Krieger, S.; Gaildrat, P.; Révillion, F.; Buisine, M.P.; Killian, A.; Duponchel, C.; Rousselin, A.; Vaur, D.; Peyrat, J.P.; et al. Contribution of bioinformatics predictions and functional splicing assays to the interpretation of unclassified variants of the BRCA genes. Eur. J. Hum. Genet. 2011, 19, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, A.Y.; Dandanell, M.; Jønson, L.; Ejlertsen, B.; Gerdes, A.M.; Nielsen, F.C.; Hansen, T. Functional characterization of BRCA1 gene variants by mini-gene splicing assay. Eur. J. Hum. Genet. 2014, 22, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Nozu, K.; Hiramoto, R.; Minamikawa, S.; Yamamura, T.; Fujimura, J.; Horinouchi, T.; Ninchoji, T.; Kaito, H.; Morisada, N.; et al. A comparison of splicing assays to detect an intronic variant of the OCRL gene in Lowe syndrome. Eur. J. Med. Genet. 2017, 60, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Paredes, F.J.; Ramos-Trujillo, E.; Claverie-Martin, F. Three exonic mutations in polycystic kidney disease-2 gene (PKD2) alter splicing of its pre-mRNA in a minigene system. Gene 2016, 578, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Hastings, M.L.; Calarco, J.A.; de Stanchina, E.; Krainer, A.R. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am. J. Hum. Genet. 2006, 78, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Kashima, T.; Rao, N.; David, C.J.; Manley, J.L. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum. Mol. Genet. 2007, 16, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Goina, E.; Skoko, N.; Pagani, F. Binding of DAZAP1 and hnRNPA1/A2 to an exonic splicing silencer in a natural BRCA1 exon 18 mutant. Mol. Cell. Biol. 2008, 28, 3850–3860. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Imaz, E.; Martín, Y.; de Conti, L.; Melean, G.; Valero, A.; Baralle, M.; Hernández-Chico, C. Functional Analysis of Mutations in Exon 9 of NF1 Reveals the Presence of Several Elements Regulating Splicing. PLoS ONE 2015, 10, e0141735. [Google Scholar] [CrossRef] [PubMed]
- Gaildrat, P.; Krieger, S.; Di Giacomo, D.; Abdat, J.; Révillion, F.; Caputo, S.; Vaur, D.; Jamard, E.; Bohers, E.; Ledemeney, D.; et al. Multiple sequence variants of BRCA2 exon 7 alter splicing regulation. J. Med. Genet. 2012, 49, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Soukarieh, O.; Gaildrat, P.; Hamieh, M.; Drouet, A.; Baert-Desurmont, S.; Frébourg, T.; Tosi, M.; Martins, A. Exonic splicing mutations are more prevalent than currently estimated and can be predicted by using in silico tools. PLoS Genet. 2016, 12, e1005756. [Google Scholar] [CrossRef]
- Veltrop, M.; Aartsma-Rus, A. Antisense-mediated exon skipping: Taking advantage of a trick from Mother Nature to treat rare genetic diseases. Exp. Cell Res. 2014, 325, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Rendu, J.; Montjean, R.; Coutton, C.; Suri, M.; Chicanne, G.; Petiot, A.; Brocard, J.; Grunwald, D.; Pietri-Rouxel, F.; Payrastre, B.; et al. Functional Characterization and Rescue of a Deep Intronic Mutation in OCRL Gene Responsible for Lowe Syndrome. Hum. Mutat. 2017, 38, 152–159. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mutation | Reference | Disease | Exon | Position 1 | FAS-ESS | ESE Finder | Rescue ESE | MutPred Splice 3 | SPANR | Splice Effect Observed | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gained ESS 2 | Disrupted ESE 2 | Result Confident Hypothesis | PSI | ||||||||
| c.725T>C p.(Phe242Ser) | [19] | Lowe S. | 9 | +3 | 0 | 0 | 0 | SAV | --- | Increased | None |
| c.741G>T p.(Trp247Cys) | [37] | Lowe S. | 9 | +19 | 0 | 0 | 2 | SAV | ESE loss, ESS gain | Decreased | Exon 9 skipping |
| c.1060A>C p.(Asn354His) | [19] | Dent-2 | 12 | +4 | 0 | 1 | 2 | SAV | --- | --- | None |
| c.1070G>T p.(Gly357Val) | [22] | Lowe S. | 12 | +14 | 0 | 0 | 0 | SAV | Cryptic 5′ SS | --- | None |
| c.1070G>A p.(Gly357Glu) | [38] | Lowe S. | 12 | +14 | 1 | 0 | 0 | SAV | ESS loss, Cryptic 5′ SS | --- | None |
| c.1221G>A p.(Pro407Pro) | This study | Dent-2 | 12 | −24 | 0 | 1 | 0 | SNV | --- | --- | None |
| c.1484C>T p.(Pro495Leu) | [39] | Lowe S. | 15 | +18 | 2 | 3 | 0 | SAV | ESE loss, ESS gain | --- | None |
| c.1489T>G p.(Trp497Gly) | [40] | Lowe S. | 15 | +23 | 2 | 0 | 0 | SAV | ESE loss, ESS gain | --- | None |
| c.1493G>A p.(Cys498Tyr) | [38] | Lowe S. | 15 | +27 | 0 | 0 | 0 | SAV | Cryptic 5′ SS | --- | None |
| c.1576C>T p.(Pro526Ser) | [41] | Dent-2 | 15 | −27 | 0 | 0 | 0 | SAV | ESE loss | --- | None |
| c.1577C>T p.(Pro526Leu) | [42] | Lowe S. | 15 | −26 | 1 | 0 | 0 | SAV | ESE loss, ESS gain | --- | None |
| c.2389G>C p.(Ala797Pro) | [38] | Lowe S. | 22 | +48 | 0 | 0 | 0 | SNV | --- | --- | None |
| c.2581G>A p.(Ala861Thr) | [11] | Lowe S. | 23 | −1 | 0 | 0 | 0 | SAV | Loss of natural 5′ SS | Decreased | Exon 23 skipping |
| c.2581G>C p.(Ala861Pro) | [22] | Lowe S. | 23 | −1 | 0 | 0 | 0 | SAV | Loss of natural 5′ SS | Decreased | Exon 23 skipping |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suarez-Artiles, L.; Perdomo-Ramirez, A.; Ramos-Trujillo, E.; Claverie-Martin, F. Splicing Analysis of Exonic OCRL Mutations Causing Lowe Syndrome or Dent-2 Disease. Genes 2018, 9, 15. https://doi.org/10.3390/genes9010015
Suarez-Artiles L, Perdomo-Ramirez A, Ramos-Trujillo E, Claverie-Martin F. Splicing Analysis of Exonic OCRL Mutations Causing Lowe Syndrome or Dent-2 Disease. Genes. 2018; 9(1):15. https://doi.org/10.3390/genes9010015
Chicago/Turabian StyleSuarez-Artiles, Lorena, Ana Perdomo-Ramirez, Elena Ramos-Trujillo, and Felix Claverie-Martin. 2018. "Splicing Analysis of Exonic OCRL Mutations Causing Lowe Syndrome or Dent-2 Disease" Genes 9, no. 1: 15. https://doi.org/10.3390/genes9010015
APA StyleSuarez-Artiles, L., Perdomo-Ramirez, A., Ramos-Trujillo, E., & Claverie-Martin, F. (2018). Splicing Analysis of Exonic OCRL Mutations Causing Lowe Syndrome or Dent-2 Disease. Genes, 9(1), 15. https://doi.org/10.3390/genes9010015