Delineation of Novel Autosomal Recessive Mutation in GJA3 and Autosomal Dominant Mutations in GJA8 in Pakistani Congenital Cataract Families

Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Clinical Findings
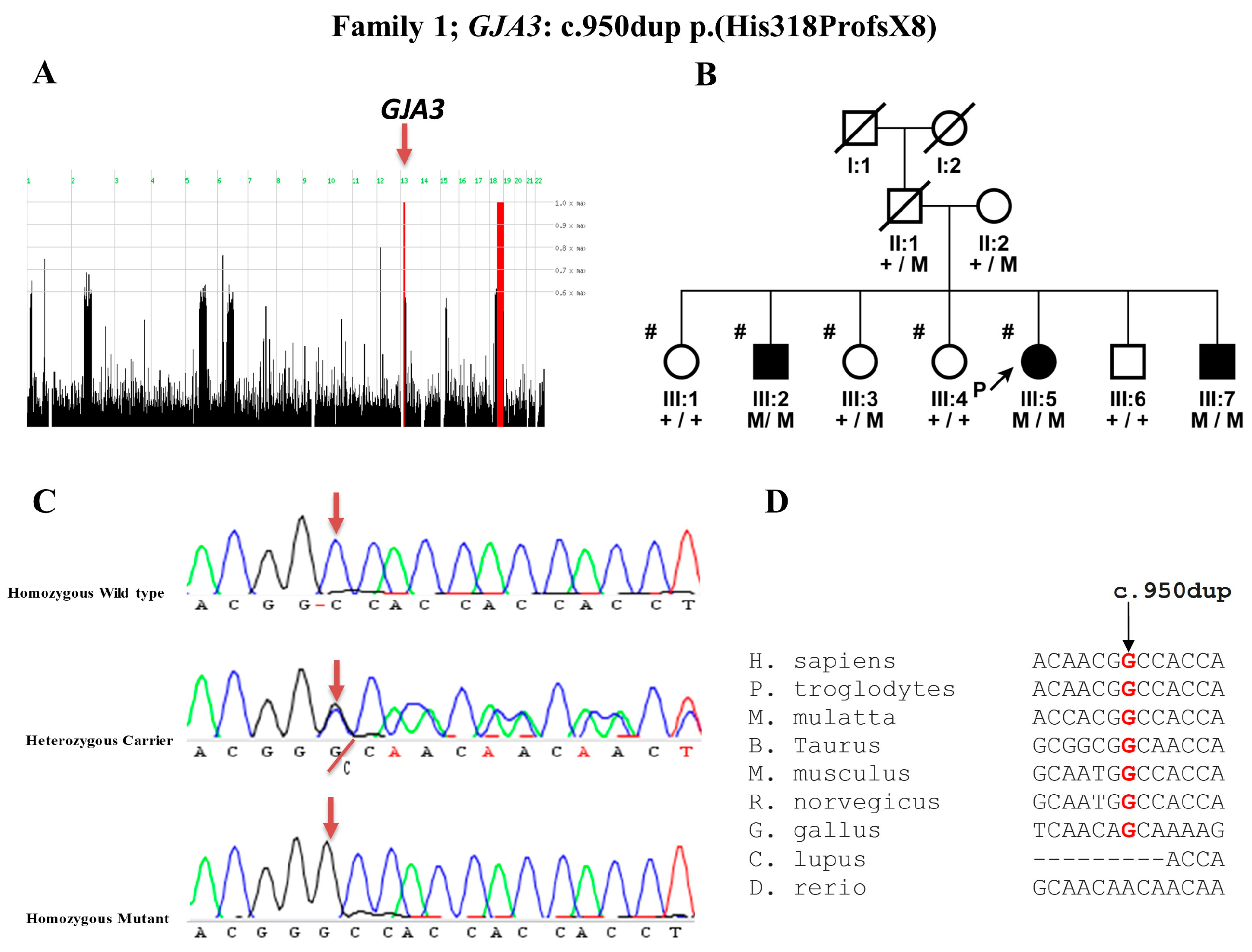
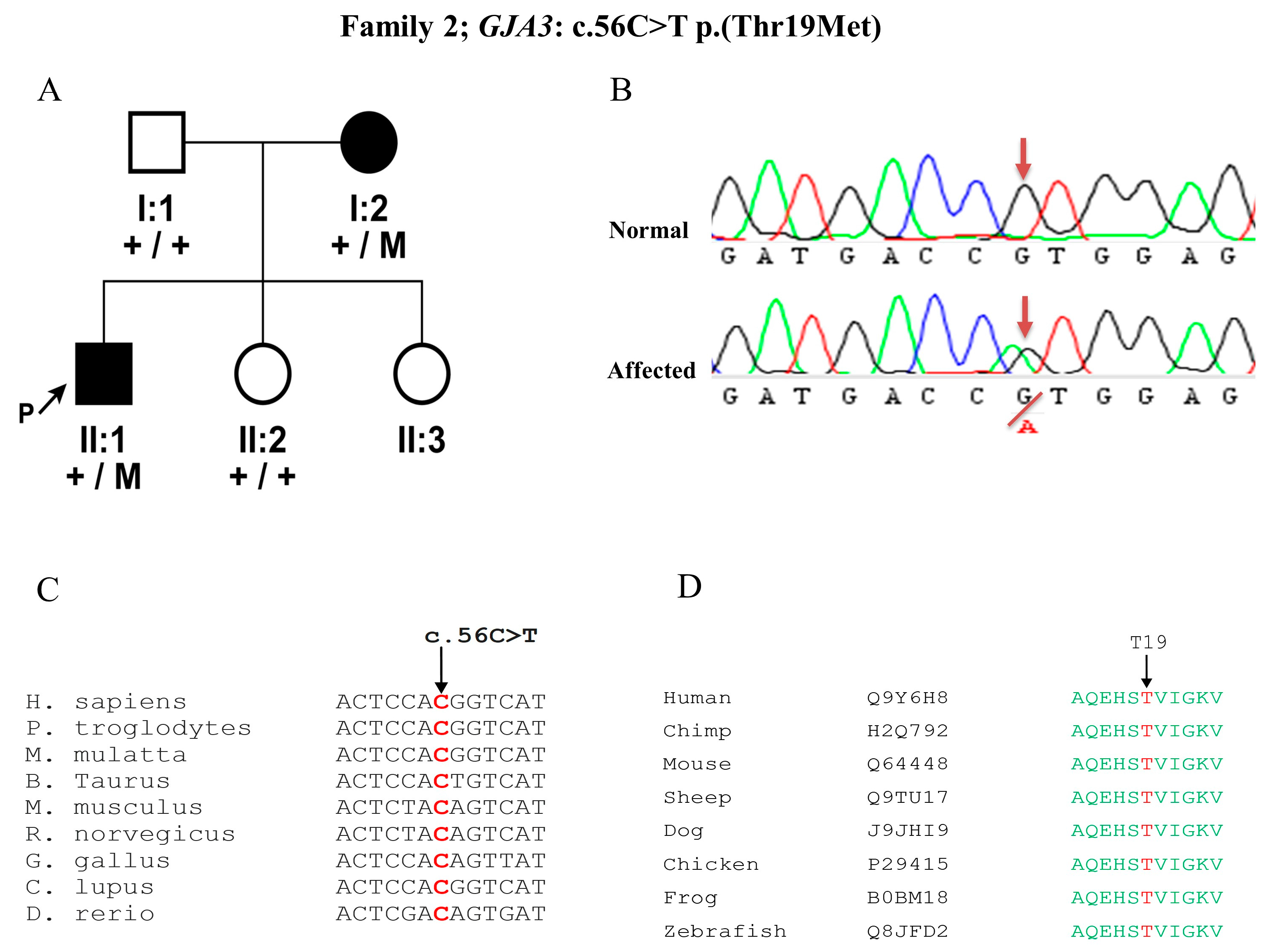
3.2. Mutation Detection in GJA3 Gene
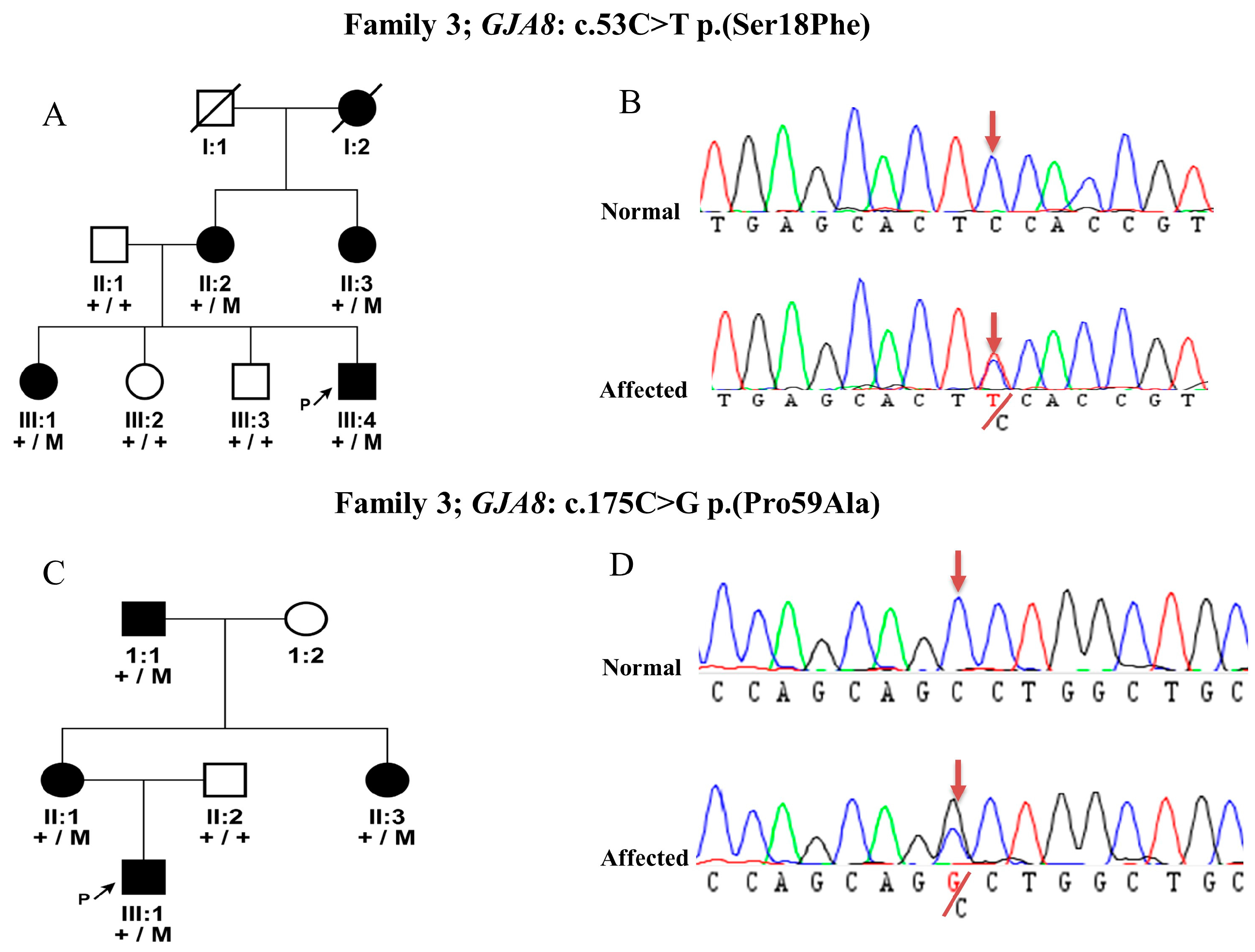
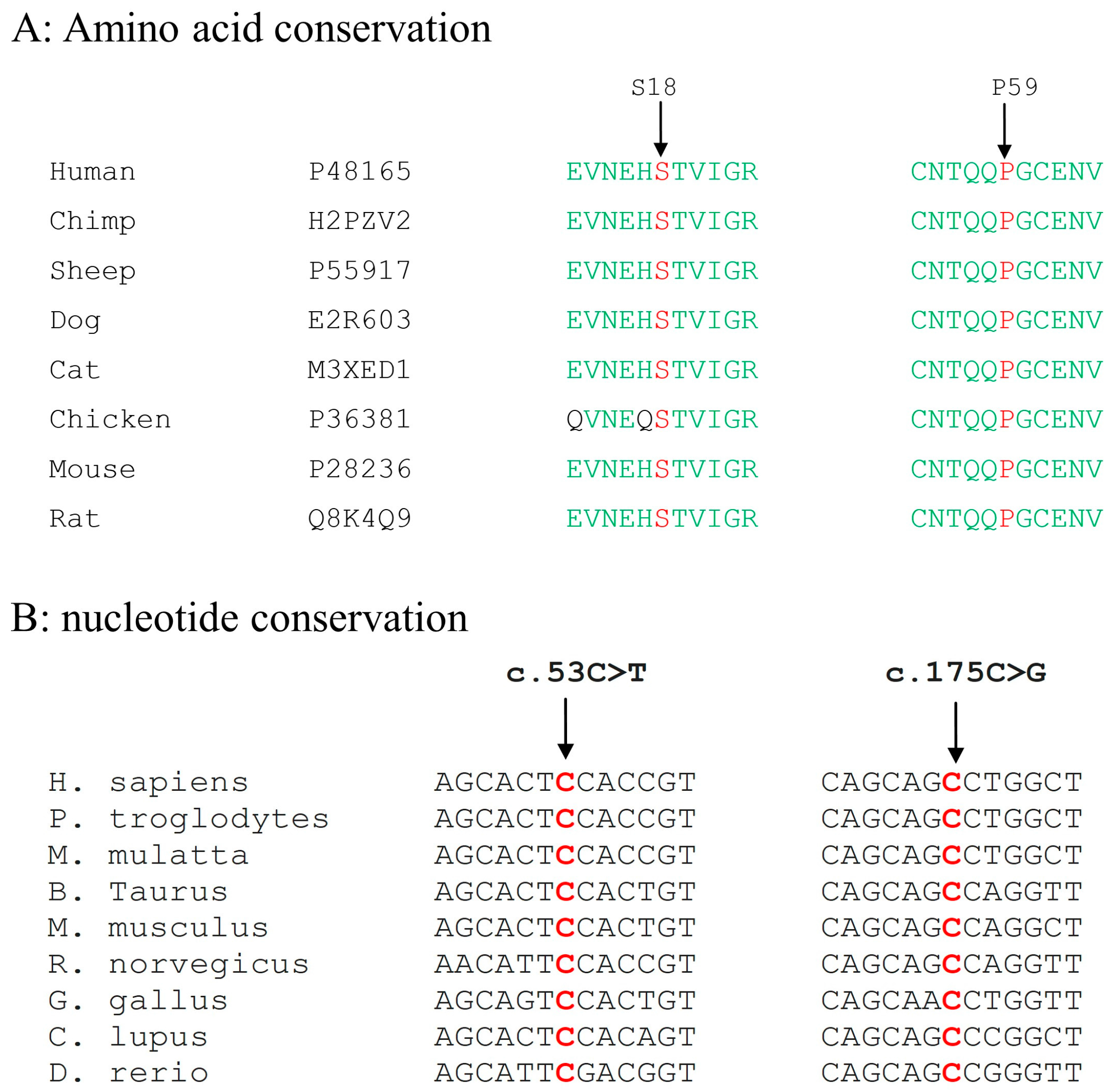
3.3. Mutation Detection in GJA8 Gene
4. Discussion
Author Contributions
Conflicts of interest
References
- Apple, D.J.; Ram, J.; Foster, A.; Peng, Q. Elimination of cataract blindness: A global perspective entering the new millenium. Surv. Ophthalmol. 2000, 45 (Suppl. 1), S1–S196. [Google Scholar] [PubMed]
- Foster, A.; Gilbert, C.; Rahi, J. Epidemiology of cataract in childhood: A global perspective. J. Cataract Refract. Surg. 1997, 23 (Suppl. 1), 601–604. [Google Scholar] [CrossRef]
- Pichi, F.; Lembo, A.; Serafino, M.; Nucci, P. Genetics of congenital cataract. Dev. Ophthalmol. 2016, 57, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xiao, W. Congenital cataract: Progress in surgical treatment and postoperative recovery of visual function. Eye Sci. 2015, 30, 38–47. [Google Scholar] [PubMed]
- Hejtmancik, J.F. Congenital cataracts and their molecular genetics. Semin. Cell Dev. Biol. 2008, 19, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Berry, V.; Francis, P.; Kaushal, S.; Moore, A.; Bhattacharya, S. Missense mutations in MIP underlie autosomal dominant ‘polymorphic’ and lamellar cataracts linked to 12q. Nat. Genet. 2000, 25, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, P.M.; Hess, J.F.; FitzGerald, P.G.; Kramer, P.; Weleber, R.G.; Litt, M. Autosomal-dominant congenital cataract associated with a deletion mutation in the human beaded filament protein gene BFSP2. Am. J. Hum. Genet. 2000, 66, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Semina, E.V.; Ferrell, R.E.; Mintz-Hittner, H.A.; Bitoun, P.; Alward, W.L.; Reiter, R.S.; Funkhauser, C.; Daack-Hirsch, S.; Murray, J.C. A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat. Genet. 1998, 19, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Vanita, V.; Singh, D.; Robinson, P.N.; Sperling, K.; Singh, J.R. A novel mutation in the DNA-binding domain of MAF at 16q23.1 associated with autosomal dominant “cerulean cataract” in an Indian family. Am. J. Med. Genet. A 2006, 140, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Behnam, M.; Imagawa, E.; Chaleshtori, A.R.; Ronasian, F.; Salehi, M.; Miyake, N.; Matsumoto, N. A novel homozygous mutation in HSF4 causing autosomal recessive congenital cataract. J. Hum. Genet. 2016, 61, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Irum, B.; Khan, S.Y.; Ali, M.; Kaul, H.; Kabir, F.; Rauf, B.; Fatima, F.; Nadeem, R. Mutation in LIM2 is responsible for autosomal recessive congenital cataracts. PLoS ONE 2016, 11, e0162620. [Google Scholar] [CrossRef] [PubMed]
- Pras, E.; Raz, J.; Yahalom, V.; Frydman, M.; Garzozi, H.J.; Pras, E.; Hejtmancik, J.F. A nonsense mutation in the glucosaminyl (N-acetyl) transferase 2 gene (GCNT2): Association with autosomal recessive congenital cataracts. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1940–1945. [Google Scholar] [CrossRef]
- Shiels, A.; Bennett, T.M.; Hejtmancik, J.F. Cat-Map: Putting cataract on the map. Mol. Vis. 2010, 16, 2007–2015. [Google Scholar] [PubMed]
- Yoshida, M.; Harada, Y.; Kaidzu, S.; Ohira, A.; Masuda, J.; Nabika, T. New genetic model rat for congenital cataracts due to a connexin 46 (GJA3) mutation. Pathol. Int. 2005, 55, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Ji, H.; Wei, Z.; Guo, L.; Li, Y.; Wang, T.; Zhu, Y.; Dong, X.; Wang, Y.; Xing, Q.; et al. A novel insertional mutation in the connexin 46 (gap junction alpha 3) gene associated with autosomal dominant congenital cataract in a Chinese family. Mol. Vis. 2013, 19, 789–795. [Google Scholar] [PubMed]
- Guleria, K.; Sperling, K.; Singh, D.; Varon, R.; Singh, J.R.; Vanita, V. A novel mutation in the connexin 46 (GJA3) gene associated with autosomal dominant congenital cataract in an Indian family. Mol. Vis. 2007, 13, 1657–1665. [Google Scholar] [PubMed]
- Pfenniger, A.; Wohlwend, A.; Kwak, B.R. Mutations in connexin genes and disease. Eur. J. Clin. Investig. 2011, 41, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef]
- Ponnam, S.P.; Ramesha, K.; Tejwani, S.; Ramamurthy, B.; Kannabiran, C. Mutation of the gap junction protein alpha 8 (GJA8) gene causes autosomal recessive cataract. J. Med. Genet. 2007, 44, e85. [Google Scholar] [CrossRef] [PubMed]
- Santhiya, S.T.; Kumar, G.S.; Sudhakar, P.; Gupta, N.; Klopp, N.; Illig, T.; Söker, T.; Groth, M.; Platzer, M.; Gopinath, P.M.; et al. Molecular analysis of cataract families in India: New mutations in the CRYBB2 and GJA3 genes and rare polymorphisms. Mol. Vis. 2010, 16, 1837–1847. [Google Scholar] [PubMed]
- Gong, X.; Li, E.; Klier, G.; Huang, Q.; Wu, Y.; Lei, H.; Kumar, N.M.; Horwitz, J.; Gilula, N.B. Disruption of alpha3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell 1997, 91, 833–843. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, M.; Chen, X.; Zhu, Y.; Gong, X.; Yao, K. Identification and functional analysis of two novel connexin 50 mutations associated with autosome dominant congenital cataracts. Sci. Rep. 2016, 6, 26551. [Google Scholar] [CrossRef] [PubMed]
- Mackay, D.S.; Bennett, T.M.; Culican, S.M.; Shiels, A. Exome sequencing identifies novel and recurrent mutations in GJA8 and CRYGD associated with inherited cataract. Hum. Genomics 2014, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.C.; Minogue, P.J.; Valiunas, V.; Kanaporis, G.; Brink, P.R.; Berthoud, V.M.; Beyer, E.C. Cataracts are caused by alterations of a critical N-terminal positive charge in connexin50. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- White, T.W. Unique and redundant connexin contributions to lens development. Science 2002, 295, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Das, S.; Molina, S.A.; Madgwick, D.; Katz, M.R.; Jena, S.; Bossmann, L.K.; Pal, D.; Takemoto, D.J. Investigation of the reciprocal relationship between the expression of two gap junction connexin proteins, connexin46 and connexin43. J. Biol. Chem. 2011, 286, 24519–24533. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, M.; Oyamada, Y.; Takamatsu, T. Regulation of connexin expression. Biochim. Biophys. Acta 2005, 1719, 6–23. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Yu, X.S.; Yin, X.; Jiang, J.X. Stimulation of lens cell differentiation by gap junction protein connexin 45.6. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2103–2111. [Google Scholar] [CrossRef]
- Shi, Q.; Jiang, J.X. Connexin arrests the cell cycle through cytosolic retention of an E3 ligase. Mol. Cell. Oncol. 2016, 3, e1132119. [Google Scholar] [CrossRef] [PubMed]
- Minogue, P.J.; Tong, J.J.; Arora, A.; Russell-Eggitt, I.; Hunt, D.M.; Moore, A.T.; Ebihara, L.; Beyer, E.C.; Berthoud, V.M. A mutant connexin50 with enhanced hemichannel function leads to cell death. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5837–5845. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.J.; Minogue, P.J.; Kobeszko, M.; Beyer, E.C.; Berthoud, V.M.; Ebihara, L. The connexin46 mutant, Cx46T19M, causes loss of gap junction function and alters hemi-channel gating. J. Membr. Biol. 2015, 248, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Riquelme, M.A.; Xu, J.; Yan, X.; Nicholson, B.J.; Gu, S.; Jiang, J. Cataract-causing mutation of human connexin 46 impairs gap junction, but increases hemichannel function and cell death. PLoS ONE 2013, 8, e74732. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C.; Berthoud, V.M. Connexin hemichannels in the lens. Front. Physiol. 2014, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Paznekas, W.A.; Boyadjiev, S.A.; Shapiro, R.E.; Daniels, O.; Wollnik, B.; Keegan, C.E.; Innis, J.W.; Dinulos, M.B.; Christian, C.; Hannibal, M.C.; et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 2003, 72, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Frasson, M.; Calixto, N.; Cronemberger, S.; de Aguiar, R.A.; Leao, L.L.; de Aguiar, M.J. Oculodentodigital dysplasia: Study of ophthalmological and clinical manifestations in three boys with probably autosomal recessive inheritance. Ophthalmic Genet. 2004, 25, 227–236. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | cDNA Position | Amino Acid Position | Study | phyloP | Grantham Score | SIFT | Mutation Taster | Poly Phen-2 |
|---|---|---|---|---|---|---|---|---|
| GJA3 | c.950dup | p.(His318ProfsX8) | Current | N/A | N/A | D | Disease causing | Damaging |
| c.56C>T | p.(Thr19Met) | Current | 6.18 | 81 | D | Disease causing | damaging | |
| c.427G>A | p.(Gly143Arg) | Previous | 5.86 | 125 | D | Disease causing | damaging | |
| c.137G>T | p.(Gly46Val) | Previous | 5.94 | 109 | D | Disease causing | damaging | |
| GJA8 | c.53C>T | p.(Ser18Phe) | Current | 5.86 | 155 | T | Disease causing | damaging |
| c.175C>G | p.(Pro59Ala) | Current | 5.96 | 27 | D | Disease causing | damaging | |
| c.20T>C | p.(Leu7Pro) | Previous | 3.35 | 98 | T | Disease causing | damaging | |
| c.68G>C | p.(Arg23Thr) | Previous | 4.16 | 71 | T | Disease causing | damaging |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micheal, S.; Niewold, I.T.G.; Siddiqui, S.N.; Zafar, S.N.; Khan, M.I.; Bergen, A.A.B. Delineation of Novel Autosomal Recessive Mutation in GJA3 and Autosomal Dominant Mutations in GJA8 in Pakistani Congenital Cataract Families. Genes 2018, 9, 112. https://doi.org/10.3390/genes9020112
Micheal S, Niewold ITG, Siddiqui SN, Zafar SN, Khan MI, Bergen AAB. Delineation of Novel Autosomal Recessive Mutation in GJA3 and Autosomal Dominant Mutations in GJA8 in Pakistani Congenital Cataract Families. Genes. 2018; 9(2):112. https://doi.org/10.3390/genes9020112
Chicago/Turabian StyleMicheal, Shazia, Ilse Therésia Gabriëla Niewold, Sorath Noorani Siddiqui, Saemah Nuzhat Zafar, Muhammad Imran Khan, and Arthur A. B. Bergen. 2018. "Delineation of Novel Autosomal Recessive Mutation in GJA3 and Autosomal Dominant Mutations in GJA8 in Pakistani Congenital Cataract Families" Genes 9, no. 2: 112. https://doi.org/10.3390/genes9020112
APA StyleMicheal, S., Niewold, I. T. G., Siddiqui, S. N., Zafar, S. N., Khan, M. I., & Bergen, A. A. B. (2018). Delineation of Novel Autosomal Recessive Mutation in GJA3 and Autosomal Dominant Mutations in GJA8 in Pakistani Congenital Cataract Families. Genes, 9(2), 112. https://doi.org/10.3390/genes9020112