Alternative Splicing of Alpha- and Beta-Synuclein Genes Plays Differential Roles in Synucleinopathies


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction—the Synuclein Family
1.1. α-Synuclein—Structure and Function
1.2. β-Synuclein—Structure and Function
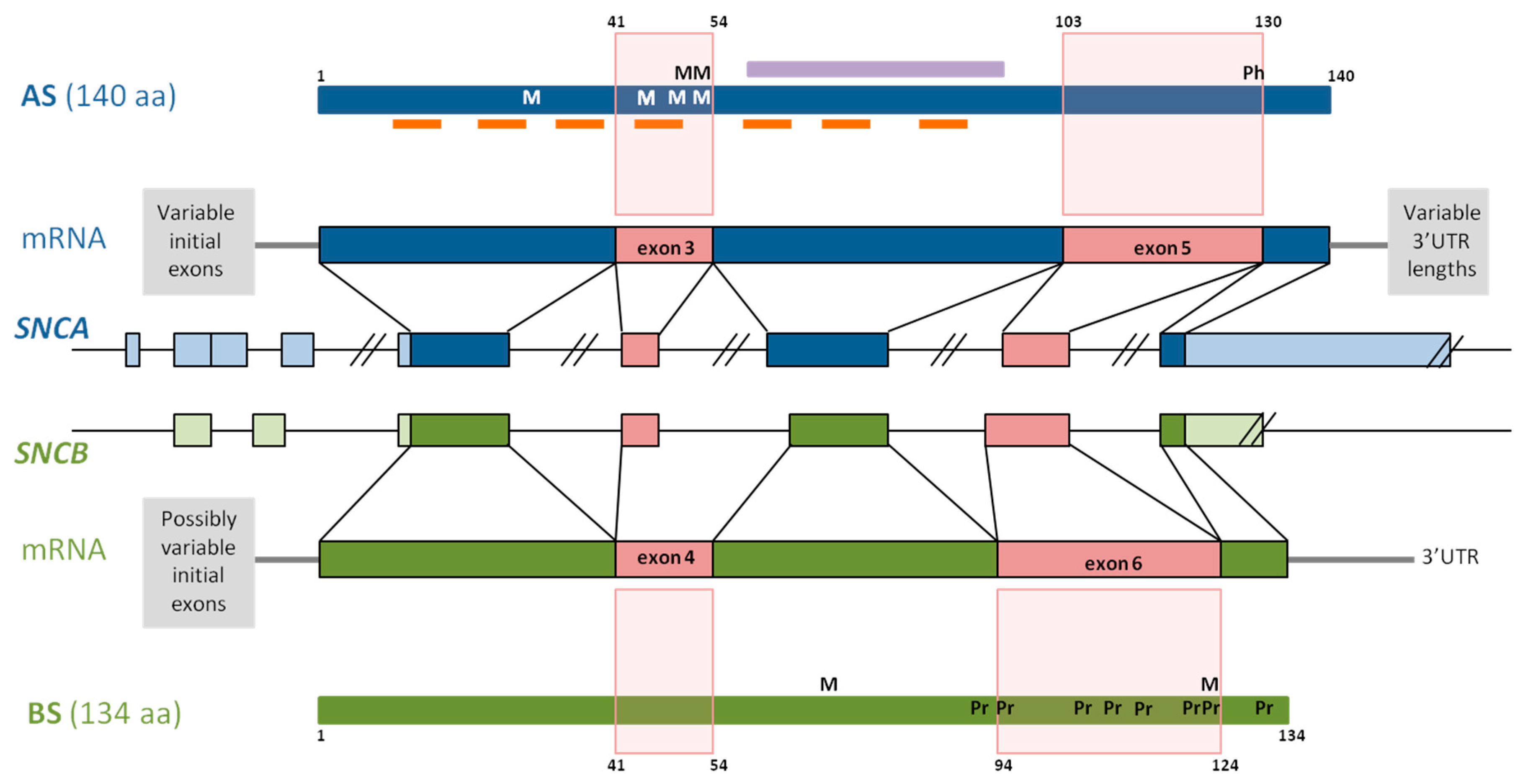
2. SNCA Alternative Splicing and Its Role in Synucleinopathies
2.1. 5’ Untranslated Region Splicing
2.2. Exon Skipping
2.2.1. In-Frame Splicing of Exon 5
2.2.2. In-Frame Splicing of Exon 3
2.2.3. In-Frame Splicing of Exons 3 and 5
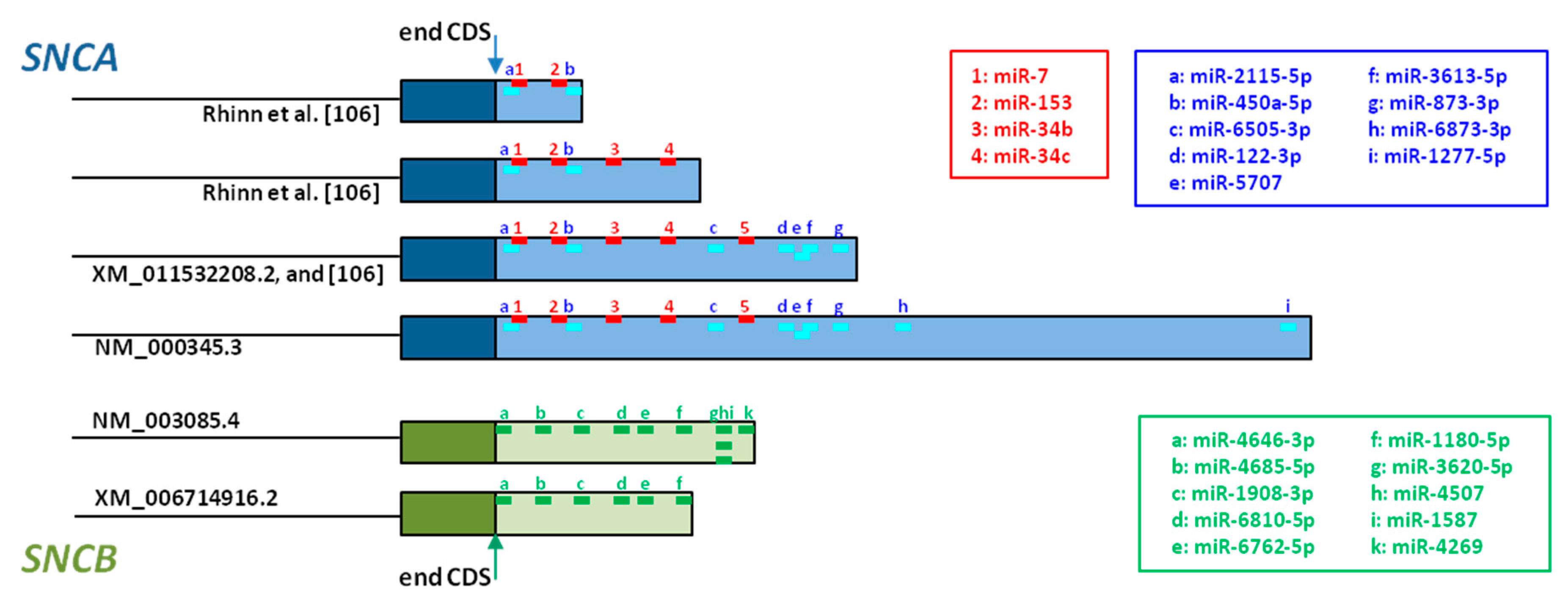
2.3. 3’ Untranslated Region Splicing
3. SNCB Alternative Splicing and Its Role in Synucleinopathies
3.1. 5’ Untranslated Region Splicing
3.2. Exon Skipping
3.3. 3’ Untranslated Region Splicing
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254. [Google Scholar] [CrossRef]
- Lücking, C.B.; Brice, A. Alpha-synuclein and Parkinson’s disease. Cell Mol. Life Sci. 2000, 57, 1894–1908. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. Synucleins: Are they two-edged swords? J. Neurosci. Res. 2013, 91, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Looking at the recent advances in understanding α-synuclein and its aggregation through the proteoform prism. F1000Research 2017, 6, 525. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of α-synuclein assembly by β- and γ-synucleins. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef] [PubMed]
- Yamin, G.; Munishkina, L.A.; Karymov, M.A.; Lyubchenko, Y.L.; Uversky, V.N.; Fink, A.L. Forcing nonamyloidogenic β-synuclein to fibrillate. Biochemistry 2005, 44, 9096–9107. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Uryu, K.; Lee, V.M.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains α-, β-, and γ-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Peters, O.; Millership, S.; Ninkina, N.; Doig, N.; Connor-Robson, N.; Threlfell, S.; Kooner, G.; Deacon, R.M.; Bannerman, D.M.; et al. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci. 2011, 31, 7264–7274. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.; Sudhof, T.C. α-synuclein promotes sSNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. αβγ-synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef] [PubMed]
- Al-Wandi, A.; Ninkina, N.; Millership, S.; Williamson, S.J.; Jones, P.A.; Buchman, V.L. Absence of α-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 2010, 31, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for α- and β-synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.C.; Schmidt, O.; Ninkina, N.; Jones, P.A.; Sharkey, J.; Buchman, V.L. Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of γ-synuclein, α-synuclein and double α/γ-synuclein null mutant mice. J. Neurochem. 2004, 89, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Connor-Robson, N.; Peters, O.M.; Millership, S.; Ninkina, N.; Buchman, V.L. Combinational losses of synucleins reveal their differential requirements for compensating age-dependent alterations in motor behavior and dopamine metabolism. Neurobiol. Aging 2016, 46, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to the rapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. The pathomechanisms underlying Parkinson’s disease. Expert Rev. Neurother. 2014, 14, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Neuropathological spectrum of synucleinopathies. Mov. Disord. 2003, 18 (Suppl. S6), S2–S12. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol. 2008, 116, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ferman, T.J.; Boeve, B.F. Dementia with Lewy bodies. Neurol. Clin. 2007, 25, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Diagnostic criteria for neuropathological assessment of Alzheimer’s disease. Neurobiol. Aging 1997, 18, S85–S88. [Google Scholar] [CrossRef]
- McKeith, I.; Mintzer, J.; Aarsland, D.; Burn, D.; Chiu, H.; Cohen-Mansfield, J.; Dickson, D.; Dubois, B.; Duda, J.E.; Feldman, H.; et al. Dementia with Lewy bodies. Lancet Neurol. 2004, 3, 19–28. [Google Scholar] [CrossRef]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Andersen, K.; Larsen, J.P.; Lolk, A.; Kragh-Sørensen, P. Prevalence and characteristics of dementia in Parkinson disease: An 8-year prospective study. Arch. Neurol. 2003, 60, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Emre, M. Dementia associated with Parkinson’s disease. Lancet Neurol. 2003, 2, 229–237. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Attems, J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 2006, 112, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzakis, M.E.; Graeber, M.B.; Gentleman, S.M.; Pearce, R.K. Striatal β-amyloid deposition in Parkinson disease with dementia. J. Neuropathol. Exp. Neurol. 2008, 67, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Striatal β-amyloid deposition in Parkinson disease with dementia. J. Neuropathol. Exp. Neurol. 2008, 67, 484–485. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, H.; Sato, T.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Differences in clinical course between dementia with Lewy bodies and Alzheimer’s disease. Eur. J. Neurol. 2009, 16, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Caballol, N.; Martí, M.J.; Tolosa, E. Cognitive dysfunction and dementia in Parkinson disease. Mov. Disord. 2007, 22, S358–S366. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M.-Y. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999, 274, 7619–7622. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [CrossRef] [PubMed]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A. Comparison of structure and dynamics of micelle-bound human α-synuclein and Parkinson disease variants. J. Biol. Chem. 2005, 280, 43179–43187. [Google Scholar] [CrossRef] [PubMed]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Weinreb, P.H.; Lansbury, P.T., Jr. The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by β-amyloid: Is NAC a common trigger or target in neurodegenerative disease? Chem. Biol. 1995, 2, 163–169. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J. Neurochem. 2007, 103, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Levitan, K.; Chereau, D.; Cohen, S.I.; Knowles, T.P.; Dobson, C.M.; Fink, A.L.; Anderson, J.P.; Goldstein, J.M.; Millhauser, G.L. Conserved C-terminal charge exerts a profound influence on the aggregation rate of α-synuclein. J. Mol. Biol. 2011, 411, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, D.; Calí, T.; Szabò, I.; Brini, M. Alpha-synuclein at the intracellular and the extracellular side: Functional and dysfunctional implications. Biol. Chem. 2017, 398, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Dev, K.K.; Hofele, K.; Barbieri, S.; Buchman, V.L.; van der Putten, H. Part II: α-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology 2003, 45, 14–44. [Google Scholar] [CrossRef]
- Breda, C.; Nugent, M.L.; Estranero, J.G.; Kyriacou, C.P.; Outeiro, T.F.; Steinert, J.R.; Giorgini, F. Rab11 modulates α-synuclein mediated defects in synaptic transmission and behaviour. Hum. Mol. Genet. 2015, 24, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.; Chieregatti, E. Mechanisms of alpha-synuclein action on neurotransmission: Cell-autonomous and non-cell autonomous role. Biomolecules 2015, 5, 865–892. [Google Scholar] [CrossRef] [PubMed]
- Schapansky, J.; Nardozzi, J.D.; LaVoie, M.J. The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson’s disease. Neuroscience 2015, 302, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Norris, K.L.; Hao, R.; Chen, L.; Lai, C.H.; Kapur, M.; Shaughnessy, P.J.; Chou, D.; Yan, J.; Taylor, J.P.; Engelender, S.; et al. Convergence of Parkin, PINK1, and α-Synuclein on Stress-induced Mitochondrial Morphological Remodeling. J. Biol. Chem. 2015, 290, 13862–13874. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, C.A.; Ancolio, K.; Checler, F. Wild-type but not Parkinson’s disease related Ala-53 → Thr mutant α-synuclein protects neuronal cells from apoptotic stimuli. J. Biol. Chem. 2000, 275, 24065–24069. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. α-synuclein misfolding and neurodegenerative diseases. Curr. Protein Pept. Sci. 2008, 9, 507–540. [Google Scholar] [CrossRef] [PubMed]
- Payton, J.E.; Perrin, R.J.; Clayton, D.F.; George, J.M. Protein-protein interactions of alpha-synuclein in brain homogenates and transfected cells. Brain Res. Mol. Brain Res. 2001, 95, 138–145. [Google Scholar] [CrossRef]
- Jin, J.; Li, G.J.; Davis, J.; Zhu, D.; Wang, Y.; Pan, C.; Zhang, J. Identification of novel proteins associated with both α-synuclein and DJ-1. Mol. Cell. Proteom. 2007, 6, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Betzer, C.; Movius, A.J.; Shi, M.; Gai, W.P.; Zhang, J.; Jensen, P.H. Identification of synaptosomal proteins binding to monomeric and oligomeric α-synuclein. PLoS ONE 2015, 10, e0116473. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Schon, E.A.; Przedborski, S. Novel subcellular localization for α-synuclein: Possible functional consequences. Front. Neuroanat. 2015, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Kermer, P.; Köhn, A.; Schnieder, M.; Lingor, P.; Bähr, M.; Liman, J.; Dohm, C.P. BAG1 is neuroprotective in in vivo and in vitro models of Parkinson’s disease. J. Mol. Neurosci. 2015, 55, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.J.; Sierecki, E.; Tomatis, V.M.; Gormal, R.S.; Giles, N.; Morrow, I.C.; Xia, D.; Götz, J.; Parton, R.G.; Collins, B.M.; et al. Munc18-1 is a molecular chaperone for α-synuclein, controlling its self-replicating aggregation. J. Cell Biol. 2016, 214, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Zaltieri, M.; Grigoletto, J.; Longhena, F.; Navarria, L.; Favero, G.; Castrezzati, S.; Colivicchi, M.A.; Della Corte, L.; Rezzani, R.; Pizzi, M.; et al. α-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J. Cell Sci. 2015, 128, 2231–2243. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. A critical evaluation of current staging of α-synuclein pathology in Lewy body disorders. Biochim. Biophys. Acta 2009, 1792, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Krebs, S.K.; Wolff, M.; Birk, G.; Hengerer, B. Seeding induced by α-synuclein oligomers provides evidence for spreading of α-synuclein pathology. J. Neurochem. 2009, 111, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological α-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, P.; Bonnet, A.M.; Débarges, B.; Lohmann, E.; Tison, F.; Pollak, P.; Agid, Y.; Dürr, A.; Brice, A. Causal relation between α-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Hofer, A.; Berg, D.; Asmus, F.; Niwar, M.; Ransmayr, G.; Riemenschneider, M.; Bonelli, S.B.; Steffelbauer, M.; Ceballos-Baumann, A.; Haussermann, P.; et al. The role of α-synuclein gene multiplications in early-onset Parkinson’s disease and dementia with Lewy bodies. J. Neural. Transm. 2005, 112, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. α-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease type pathology. Neurobiol. Aging 2014, 35, 2180.e1-5. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ma, B.; Nussinov, R.; Thompson, D. Familial Mutations May Switch Conformational Preferences in α-Synuclein Fibrils. ACS Chem. Neurosci. 2017, 8, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.Y. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [PubMed]
- Rivers, R.C.; Kumita, J.R.; Tartaglia, G.G.; Dedmon, M.M.; Pawar, A.; Vendruscolo, M.; Dobson, C.M.; Christodoulou, J. Molecular determinants of the aggregation behavior of α- and β-synuclein. Protein Sci. 2008, 17, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Zibaee, S.; Jakes, R.; Fraser, G.; Serpell, L.C.; Crowther, R.A.; Goedert, M. Sequence determinants for amyloid fibrillogenesis of human α-synuclein. J. Mol. Biol. 2007, 374, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Roodveldt, C.; Andersson, A.; De Genst, E.J.; Labrador-Garrido, A.; Buell, A.K.; Dobson, C.M.; Tartaglia, G.G.; Vendruscolo, M. A rationally designed six-residue swap generates comparability in the aggregation behavior of α-synuclein and β-synuclein. Biochemistry 2012, 51, 8771–8778. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Rasia, R.M.; Lamberto, G.R.; Binolfi, A.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Structural characterization of the intrinsically unfolded protein β-synuclein, a natural negative regulator of α-synuclein aggregation. J. Mol. Biol. 2007, 372, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, A.A.; Sternberg, M.J.; Makarov, A.A. Polyproline-II helix in proteins: Structure and function. J. Mol. Biol. 2013, 425, 2100–2132. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Sugama, S.; Sekiyama, K.; Sekigawa, A.; Tsukui, T.; Nakai, M.; Waragai, M.; Takenouchi, T.; Takamatsu, Y.; Wei, J.; et al. A β-synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat. Commun. 2010, 1, 110. [Google Scholar] [CrossRef] [PubMed]
- Vigneswara, V.; Cass, S.; Wayne, D.; Bolt, E.L.; Ray, D.E.; Carter, W.G. Molecular ageing of alpha- and Beta-synucleins: Protein damage and repair mechanisms. PLoS ONE 2013, 8, e61442. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Lansbury, P.T., Jr. β-synuclein inhibits formation of α-synuclein protofibrils: A possible therapeutic strategy against Parkinson’s disease. Biochemistry 2003, 42, 3696–3700. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Sorensen, E.S.; Petersen, T.E.; Gliemann, J.; Rasmussen, L.K. Residues in the synuclein consensus motif of the α-synuclein fragment, NAC, participate in transglutaminase-catalysed cross-linking to Alzheimer disease amyloid βA4 peptide. Biochem. J. 1995, 310, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Mallory, M.; Masliah, E. β-synuclein inhibits α-synuclein aggregation: A possible role as an anti parkinsonian factor. Neuron 2001, 32, 213–223. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Bar-On, P.; Sharikov, Y.; Crews, L.; Hashimoto, M.; Miller, M.A.; Keller, S.H.; Platoshyn, O.; Yuan, J.X.; Masliah, E. Dynamics of α-synuclein aggregation and inhibition of pore-like oligomer development by β-synuclein. FEBS J. 2007, 274, 1862–1877. [Google Scholar] [CrossRef] [PubMed]
- Israeli, E.; Sharon, R. β-synuclein occurs in vivo in lipid-associated oligomers and forms hetero-oligomers with α-synuclein. J. Neurochem. 2009, 108, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Windisch, M.; Hutter-Paier, B.; Schreiner, E.; Wronski, R. β-Synuclein derived peptides with neuroprotective activity: An alternative treatment of neurodegenerative disorders? J. Mol. Neurosci. 2004, 24, 155–165. [Google Scholar] [CrossRef]
- Shaltiel-Karyo, R.; Frenkel-Pinter, M.; Egoz-Matia, N.; Frydman-Marom, A.; Shalev, D.E.; Segal, D.; Gazit, E. Inhibiting α-synuclein oligomerization by stable cell-penetrating β-synuclein fragments recovers phenotype of Parkinson’s disease model flies. PLoS ONE 2010, 5, e13863. [Google Scholar] [CrossRef] [PubMed]
- Taschenberger, G.; Toloe, J.; Tereshchenko, J.; Akerboom, J.; Wales, P.; Benz, R.; Becker, S.; Outeiro, T.F.; Looger, L.L.; Bähr, M.; et al. β-synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Ann. Neurol. 2013, 74, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Rosado-Ramos, R.; Gerhardt, E.; Favretto, F.; Magalhães, F.; Popova, B.; Becker, S.; Zweckstetter, M.; Braus, G.H.; Outeiro, T.F. Yeast reveals similar molecular mechanisms underlying alpha- and beta-synuclein toxicity. Hum. Mol. Genet. 2016, 25, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, G.M.; Olson, M.P.; Atieh, T.B.; Janowska, M.K.; Khare, S.D.; Baum, J. A pH-dependent switch promotes β-synuclein fibril formation via glutamate residues. J. Biol. Chem. 2017, 292, 16368–16379. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, H.; Limprasert, P.; Fan, Y.; Onodera, O.; Kakita, A.; Takahashi, H.; Bonner, L.T.; Tsuang, D.W.; Murray, I.V.; Lee, V.M.; et al. β-synuclein gene alterations in dementia with Lewy bodies. Neurology 2004, 63, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Janowska, M.K.; Baum, J. The loss of inhibitory C-terminal conformations in disease associated P123H β-synuclein. Protein Sci. 2016, 25, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fujita, M.; Nakai, M.; Waragai, M.; Watabe, K.; Akatsu, H.; Rockenstein, E.; Masliah, E.; Hashimoto, M. Enhanced lysosomal pathology caused by β-synuclein mutants linked to dementia with Lewy bodies. J. Biol. Chem. 2007, 282, 28904–28914. [Google Scholar] [CrossRef] [PubMed]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Modrek, B.; Lee, C.J. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat. Genet. 2003, 34, 177–180. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Ariza, A. α-Synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol. Neurobiol. 2013, 47, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, H.; Qiang, L.; Yamashita, T.; Rhee, D.; Zolin, A.; Vanti, W.; Abeliovich, A. Alternative α-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat. Commun. 2012, 3, 1084. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. SNCA synuclein alpha [Homo sapiens (human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/6622 (accessed on 15 January 2018).
- European Bioinformatics Institute. Tools & Databases. Available online: https://www.ebi.ac.uk/Tools/ (accessed on 15 January 2018).
- Tsunoda, T.; Takagi, T. Estimating Transcription Factor Bindability on DNA. Bioinformatics 1999, 15, 622–630. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Zdhhc12 zinc finger, DHHC domain containing 12 [Mus musculus (house mouse)]. Available online: https://www.ncbi.nlm.nih.gov/gene/66220 (accessed on 15 January 2018).
- Hughes, T.A. Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006, 22, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Scherzer, C.R.; Grass, J.A.; Liao, Z.; Pepivani, I.; Zheng, B.; Eklund, A.C.; Ney, P.A.; Ng, J.; McGoldrick, M.; Mollenhauer, B.; et al. GATA transcription factors directly regulate the Parkinson’s disease-linked gene α-synuclein. Proc. Natl. Acad. Sci. USA 2008, 105, 10907–10912. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boni, L.; Tierling, S.; Roeber, S.; Walter, J.; Giese, A.; Kretzschmar, H.A. Next-generation sequencing reveals regional differences of the α-synuclein methylation state independent of Lewy body disease. Neuromol. Med. 2011, 13, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.Y.; Wu, L.; Zhao, Z.B.; Wang, Y.; Xiao, Q.; Liu, J.; Wang, G.; Ma, J.F.; Chen, S.D. Methylation of α-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Park. Relat. Disord. 2014, 20, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, I.; Kaut, O.; Khazneh, H.; deBoni, L.; Ahmad, A.; Berg, D.; Klein, C.; Fröhlich, H.; Wüllner, U. L-dopa increases α-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro. Mov. Disord. 2015, 30, 1794–17801. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Yoshino, Y.; Yamazaki, K.; Mori, Y.; Mori, T.; Ozaki, Y.; Sao, T.; Ochi, S.; Iga, J.I.; Ueno, S.I. DNA methylation changes at SNCA intron 1 in patients with dementia with Lewy bodies. Psychiatry Clin. Neurosci. 2017, 71, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Touchman, J.W.; Dehejia, A.; Chiba-Falek, O.; Cabin, D.E.; Schwartz, J.R.; Orrison, B.M.; Polymeropoulos, M.H.; Nussbaum, R.L. Human and mouse α-synuclein genes: Comparative genomic sequence analysis and identification of a novel gene regulatory element. Genome Res. 2001, 11, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Cronin, K.D.; Ge, D.; Manninger, P.; Linnertz, C.; Rossoshek, A.; Orrison, B.M.; Bernard, D.J.; El-Agnaf, O.M.; Schlossmacher, M.G.; Nussbaum, R.L.; et al. Expansion of the Parkinson disease-associated SNCA-Rep1 allele upregulates human α-synuclein in transgenic mouse brain. Hum. Mol. Genet. 2009, 18, 3274–3285. [Google Scholar] [CrossRef] [PubMed]
- Chiba-Falek, O.; Nussbaum, R.L. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum. Mol. Genet. 2001, 10, 3101–3109. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.; Maraganore, D.M.; Lockhart, P.; Singleton, A.; Lesnick, T.G.; de Andrade, M.; West, A.; de Silva, R.; Hardy, J.; Hernandez, D. Alpha-synuclein gene haplotypes are associated with Parkinson’s disease. Hum. Mol. Genet. 2001, 10, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Maraganore, D.M.; de Andrade, M.; Elbaz, A.; Farrer, M.J.; Ioannidis, J.P.; Krüger, R.; Rocca, W.A.; Schneider, N.K.; Lesnick, T.G.; Lincoln, S.J.; et al. Collaborative analysis of α-synuclein gene promoter variability and Parkinson disease. JAMA 2006, 296, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Lareau, L.F.; Brooks, A.N.; Soergel, D.A.; Meng, Q.; Brenner, S.E. The coupling of alternative splicing and nonsense-mediated mRNA decay. Adv. Exp. Med. Biol. 2007, 623, 190–211. [Google Scholar] [PubMed]
- Lewis, B.P.; Green, R.E.; Brenner, S.E. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. USA 2003, 100, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K. Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 2006, 112, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Domingo-Sábat, M.; Lao, J.I.; Carrato, C.; Ferrer, I.; Ariza, A. Identification and characterization of a new alpha-synuclein isoform and its role in Lewy body diseases. Neurogenetics 2008, 9, 15–23. [Google Scholar] [CrossRef] [PubMed]
- McClendon, S.; Rospigliosi, C.C.; Eliezer, D. Charge neutralization and collapse of the C-terminal tail of alpha-synuclein at low pH. Protein Sci. 2009, 18, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Han, J.; Zhang, C.; Ma, Q.L.; Li, X.; Cheng, F.; Liu, G.; Li, Y.; Uéda, K.; Chan, P.; et al. C-terminal part of α-synuclein mediates its activity in promoting proliferation of dopaminergic cells. J. Neural. Transm. 2011, 118, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Jao, C.C.; Der-Sarkissian, A.; Chen, J.; Langen, R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. USA 2004, 101, 8331–8336. [Google Scholar] [CrossRef] [PubMed]
- Heise, H.; Hoyer, W.; Becker, S.; Andronesi, O.C.; Riedel, D.; Baldus, M. Molecular-level secondary structure, polymorphism, and dynamics of full-length α-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. USA 2005, 102, 15871–15876. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.V.; Giasson, B.I.; Quinn, S.M.; Koppaka, V.; Axelsen, P.H.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 2003, 42, 8530–8540. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A. Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade? J. Park. Dis. 2016, 6, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Bungeroth, M.; Appenzeller, S.; Regulin, A.; Völker, W.; Lorenzen, I.; Grötzinger, J.; Pendziwiat, M.; Kuhlenbäumer, G. Differential aggregation properties of alpha-synuclein isoforms. Neurobiol. Aging 2014, 35, 1913–1919. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, J.S.; Termine, D.J.; Lindquist, S. Splice isoform and pharmacological studies reveal that sterol depletion relocalizes α-synuclein and enhances its toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 3014–3019. [Google Scholar] [CrossRef] [PubMed]
- Kalivendi, S.V.; Yedlapudi, D.; Hillard, C.J.; Kalyanaraman, B. Oxidants induce alternative splicing of α-synuclein: Implications for Parkinson’s disease. Free Radic. Biol. Med. 2010, 48, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Manda, K.M.; Yedlapudi, D.; Korukonda, S.; Bojja, S.; Kalivendi, S.V. The chaperone-like activity of α-synuclein attenuates aggregation of its alternatively spliced isoform, 112-synuclein in vitro: Plausible cross-talk between isoforms in protein aggregation. PLoS ONE 2014, 9, e98657. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; Paik, S.R.; Yang, C.H.; Kim, J. Structural changes in α-synuclein affect its chaperone-like activity in vitro. Protein Sci. 2000, 9, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; Paik, S.R.; Yang, C.H. Structural and functional implications of C-terminal regions of α-synuclein. Biochemistry 2002, 41, 13782–13790. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; McGeer, P.L. Complement activation by islet amyloid polypeptide (IAPP) and α-synuclein 112. Biochem. Biophys. Res. Commun. 2007, 357, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, D.A.; Camp, D.M.; Conant, S.B. Complement activation in the Parkinson’s disease substantia nigra: An immunocytochemical study. J. Neuroinflamm. 2006, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Inflammation and neurodegeneration in Parkinson’s disease. Park. Relat. Disord. 2004, 10 (Suppl. S1), S3–S7. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Lao, J.I.; Carrato, C.; Mate, J.L.; López, D.; Ferrer, I.; Ariza, A. Differential expression of α-synuclein isoforms in dementia with Lewy bodies. Neuropathol. Appl. Neurobiol. 2004, 30, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Domingo-Sàbat, M.; Humbert, J.; Carrato, C.; Ferrer, I.; Ariza, A. Differential expression of alpha-synuclein, parkin, and synphilin-1 isoforms in Lewy body disease. Neurogenetics 2008, 9, 163–172. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.R.; Hallett, P.J.; Cooper, O.; Stanley, M.; Isacson, O. Transcript expression levels of full-length alpha-synuclein and its three alternatively spliced variants in Parkinson’s disease brain regions and in a transgenic mouse model of alpha-synuclein overexpression. Mol. Cell. Neurosci. 2012, 49, 23230–23239. [Google Scholar] [CrossRef] [PubMed]
- Brudek, T.; Winge, K.; Rasmussen, N.B.; Bahl, J.M.; Tanassi, J.; Agander, T.K.; Hyde, T.M.; Pakkenberg, B. Altered α-synuclein, parkin, and synphilin isoform levels in multiple system atrophy brains. J. Neurochem. 2016, 136, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A.; Daniel, S.E.; Goedert, M. Characterisation of isolated αa-synuclein filaments from substantia nigra of Parkinson’s disease brain. Neurosci. Lett. 2000, 292, 128–130. [Google Scholar] [CrossRef]
- Croisier, E.; MRes, D.E.; Deprez, M.; Goldring, K.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B.; Roncaroli, F. Comparative study of commercially available anti-α-synuclein antibodies. Neuropathol. Appl. Neurobiol. 2006, 32, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.; McLean, C.A.; Culvenor, J.G.; Gai, W.P.; Blumbergs, P.C.; Jäkälä, P.; Beyreuther, K.; Masters, C.L.; Li, Q.X. The solubility of α-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J. Neurochem. 2001, 76, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Woerman, A.L.; Stöhr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef] [PubMed]
- Locascio, J.J.; Eberly, S.; Liao, Z.; Liu, G.; Hoesing, A.N.; Duong, K.; Trisini-Lipsanopoulos, A.; Dhima, K.; Hung, A.Y.; Flaherty, A.W.; et al. Association between α-synuclein blood transcripts and early, neuroimaging-supported Parkinson’s disease. Brain 2015, 138, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J.; Linnertz, C.; Saucier, L.; Burke, J.R.; Hulette, C.M.; Welsh-Bohmer, K.A.; Chiba-Falek, O. The effect of SNCA 3′ region on the levels of SNCA-112 splicing variant. Neurogenetics 2011, 12, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Beyer, K.; Humbert, J.; Ferrer, A.; Lao, J.I.; Latorre, P.; Lopez, D.; Tolosa, E.; Ferrer, I.; Ariza, A. A variable poly-T sequence modulates α-synuclein isoform expression and is associated with aging. J. Neurosci. Res. 2007, 85, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.L.; Yuan, Y.H.; Song, L.K.; Han, N.; Chen, N.H. Over-expression of α-synuclein 98 triggers intracellular oxidative stress and enhances susceptibility to rotenone. Neurosci. Lett. 2011, 491, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Ule, A.; Spencer, J.; Williams, A.; Hu, J.S.; Cline, M.; Wang, H.; Clark, T.; Fraser, C.; Ruggiu, M.; et al. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005, 37, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.B.; Dredge, B.K.; Stefani, G.; Zhong, R.; Buckanovich, R.J.; Okano, H.J.; Yang, Y.Y.; Darnell, R.B. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 2000, 25, 359–371. [Google Scholar] [CrossRef]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed]
- An, J.J.; Gharami, K.; Liao, G.Y.; Woo, N.H.; Lau, A.G.; Vanevski, F.; Torre, E.R.; Jones, K.R.; Feng, Y.; Lu, B.; et al. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 2008, 134, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Flavell, S.W.; Kim, T.K.; Gray, J.M.; Harmin, D.A.; Hemberg, M.; Hong, E.J.; Markenscoff-Papadimitriou, E.; Bear, D.M.; Greenberg, M.E. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 2008, 60, 1022–1038. [Google Scholar] [CrossRef] [PubMed]
- Flavell, S.W.; Cowan, C.W.; Kim, T.K.; Greer, P.L.; Lin, Y.; Paradis, S.; Griffith, E.C.; Hu, L.S.; Chen, C.; Greenberg, M.E. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 2006, 311, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J. From birth to death: The complex lives of eukaryotic mRNAs. Science 2005, 309, 1514–1518. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.S.; Bhattacharyya, S.N.; Artus, C.G.; Zoller, T.; Cougot, N.; Basyuk, E.; Bertrand, E.; Filipowicz, W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 2005, 309, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Yekta, S.; Shih, I.H.; Bartel, D.P. MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004, 304, 594–596. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, H.O.; Tomari, Y. The Functions of MicroRNAs: MRNA Decay and Translational Repression. Trends Cell Biol. 2015, 25, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.W.; Fletcher, S.; Wilton, S.D. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell. Mol. Life Sci. 2012, 69, 3613–3634. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Perier, C.; Sue, C.M. Role of microRNAs in the Regulation of α-Synuclein Expression: A Systematic Review. Front. Mol. Neurosci. 2016, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of α-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- Doxakis, E. Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [PubMed]
- Fragkouli, A.; Doxakis, E. miR-7 and miR-153 protect neurons against MPP+-induced cell death via upregulation of mTOR pathway. Front. Cell. Neurosci. 2014, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson’s disease. FEBS Lett. 2015, 589, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Zhang, J.L.; Duan, Y.L.; Zhang, Q.S.; Li, G.F.; Zheng, D.L. MicroRNA-214 participates in the neuroprotective effect of Resveratrol via inhibiting α-synuclein expression in MPTP-induced Parkinson’s disease mouse. Biomed. Pharmacother. 2015, 74, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRbase: microRNA sequences, targets and gene nomenclature. Available online: http://www.mirbase.org/search.shtml (accessed on 15 January 2018).
- Wong, N.; Wang, X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Hansen, L.A.; Mallory, M.; Trojanowski, J.Q.; Galasko, D.; Masliah, E. Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res. 2001, 914, 48–56. [Google Scholar] [CrossRef]
- Maroteaux, L.; Scheller, R.H. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res. Mol. Brain Res. 1991, 11, 335–343. [Google Scholar] [CrossRef]
- Beyer, K.; Domingo-Sàbat, M.; Santos, C.; Tolosa, E.; Ferrer, I.; Ariza, A. The decrease of β-synuclein in cortical brain areas defines a molecular subgroup of dementia with Lewy bodies. Brain 2010, 133, 3724–3733. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Ispierto, L.; Latorre, P.; Tolosa, E.; Ariza, A. Alpha- and beta-synuclein expression in Parkinson disease with and without dementia. J. Neurol. Sci. 2011, 310, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Munoz-Marmol, A.M.; Sanz, C.; Marginet-Flinch, R.; Ferrer, I.; Ariza, A. New brain-specific beta-synuclein isoforms show expression ratio changes in Lewy body diseases. Neurogenetics 2012, 13, 61–72. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gámez-Valero, A.; Beyer, K. Alternative Splicing of Alpha- and Beta-Synuclein Genes Plays Differential Roles in Synucleinopathies. Genes 2018, 9, 63. https://doi.org/10.3390/genes9020063
Gámez-Valero A, Beyer K. Alternative Splicing of Alpha- and Beta-Synuclein Genes Plays Differential Roles in Synucleinopathies. Genes. 2018; 9(2):63. https://doi.org/10.3390/genes9020063
Chicago/Turabian StyleGámez-Valero, Ana, and Katrin Beyer. 2018. "Alternative Splicing of Alpha- and Beta-Synuclein Genes Plays Differential Roles in Synucleinopathies" Genes 9, no. 2: 63. https://doi.org/10.3390/genes9020063
APA StyleGámez-Valero, A., & Beyer, K. (2018). Alternative Splicing of Alpha- and Beta-Synuclein Genes Plays Differential Roles in Synucleinopathies. Genes, 9(2), 63. https://doi.org/10.3390/genes9020063