lncRNA Gene Signatures for Prediction of Breast Cancer Intrinsic Subtypes and Prognosis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sources and Description
2.2. Supervised Gene Selection Using Recursive 1-Norm SVM Method
2.3. Breast Cancer Subtype Classification and Prediction Evaluation
2.4. Prognosis Evaluation for Selected Gene Signature
3. Results
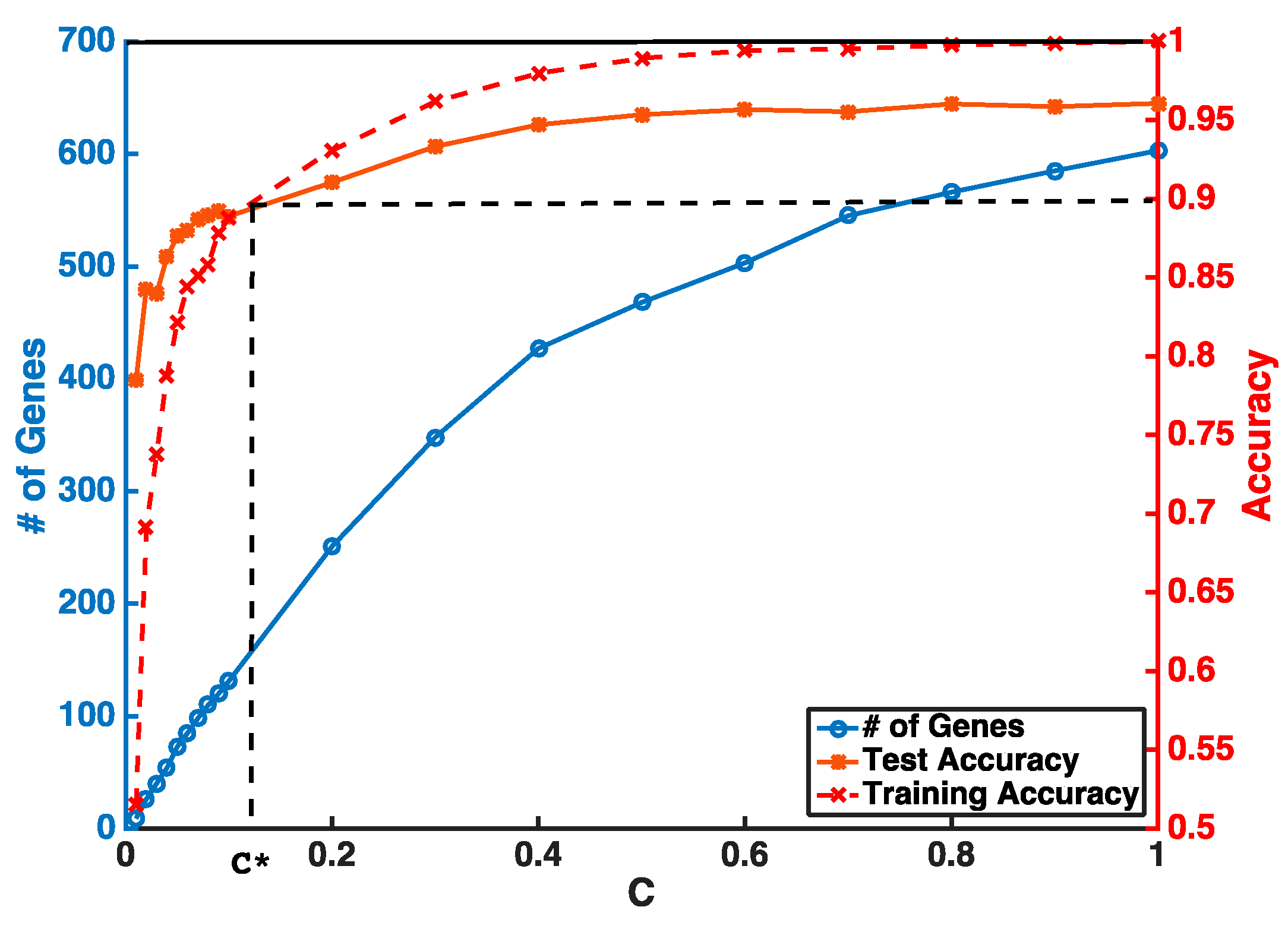
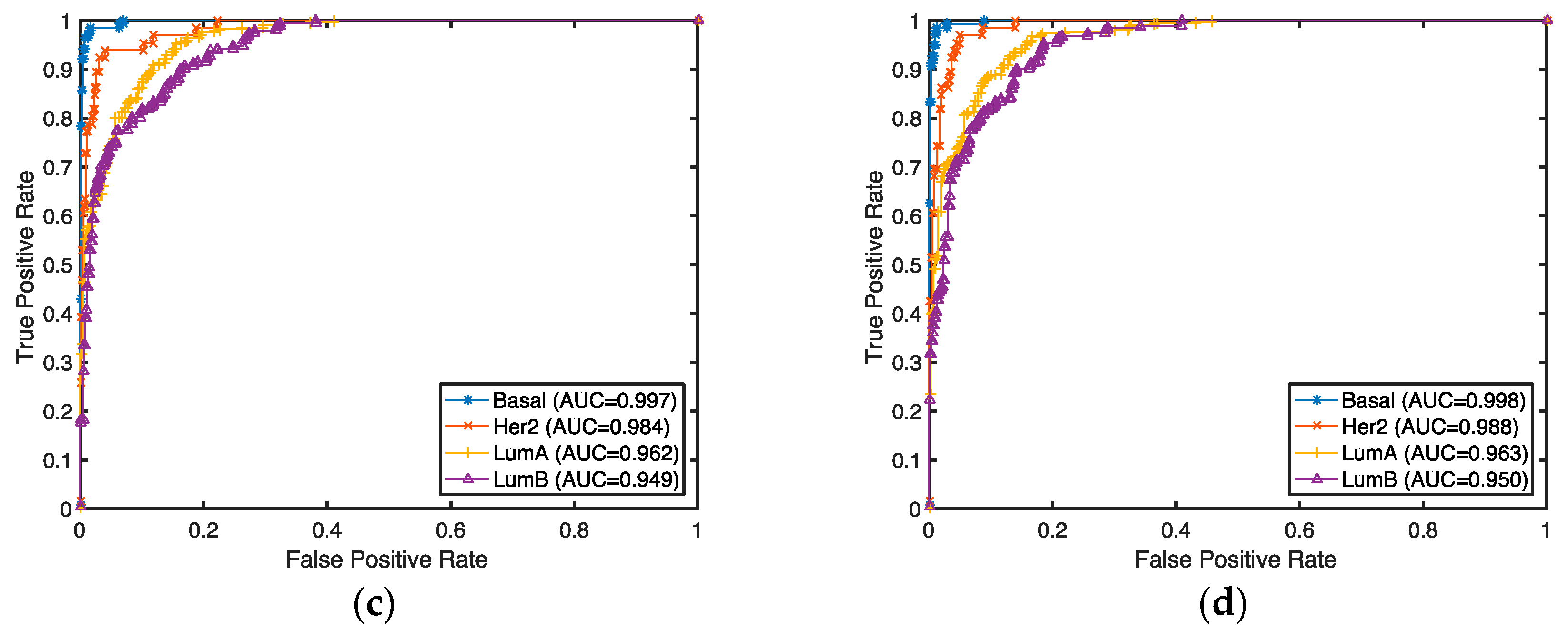
3.1. Gene Selection and Breast Cancer Subtype Classification
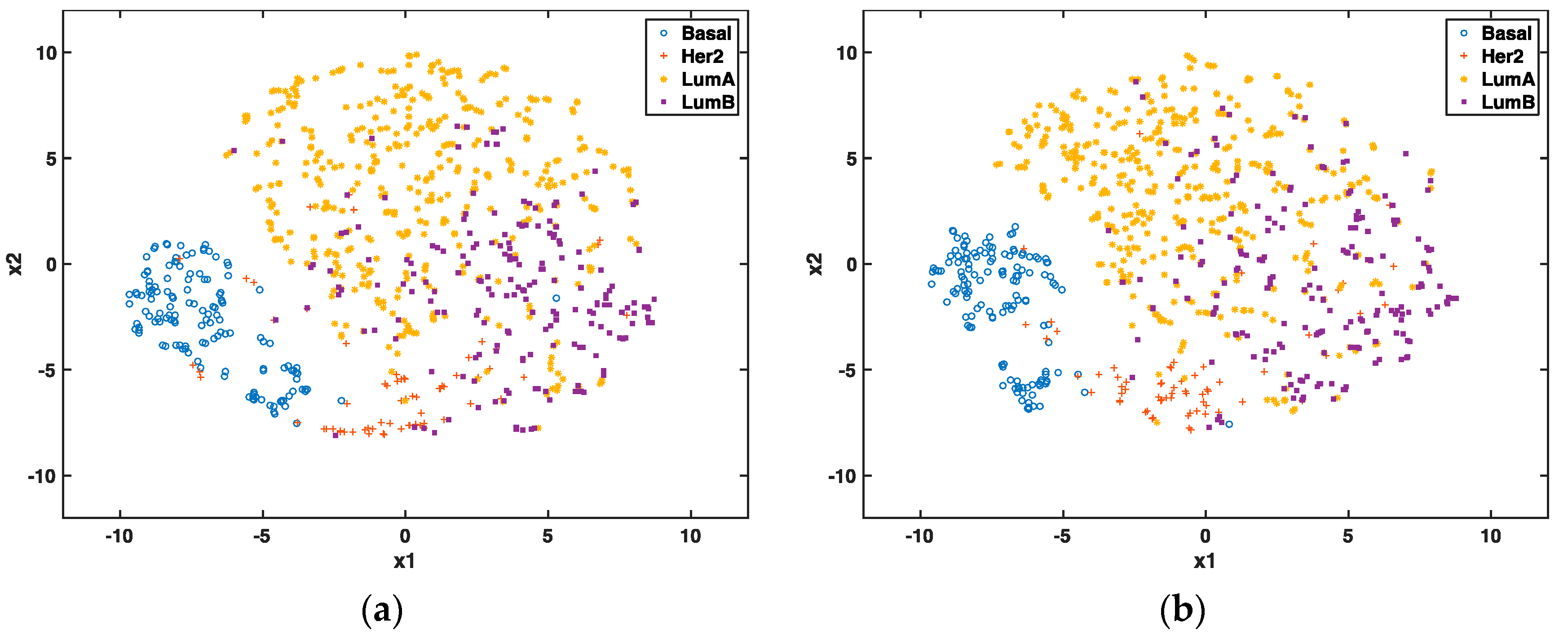
3.2. Visualization of Breast Cancer Subtypes
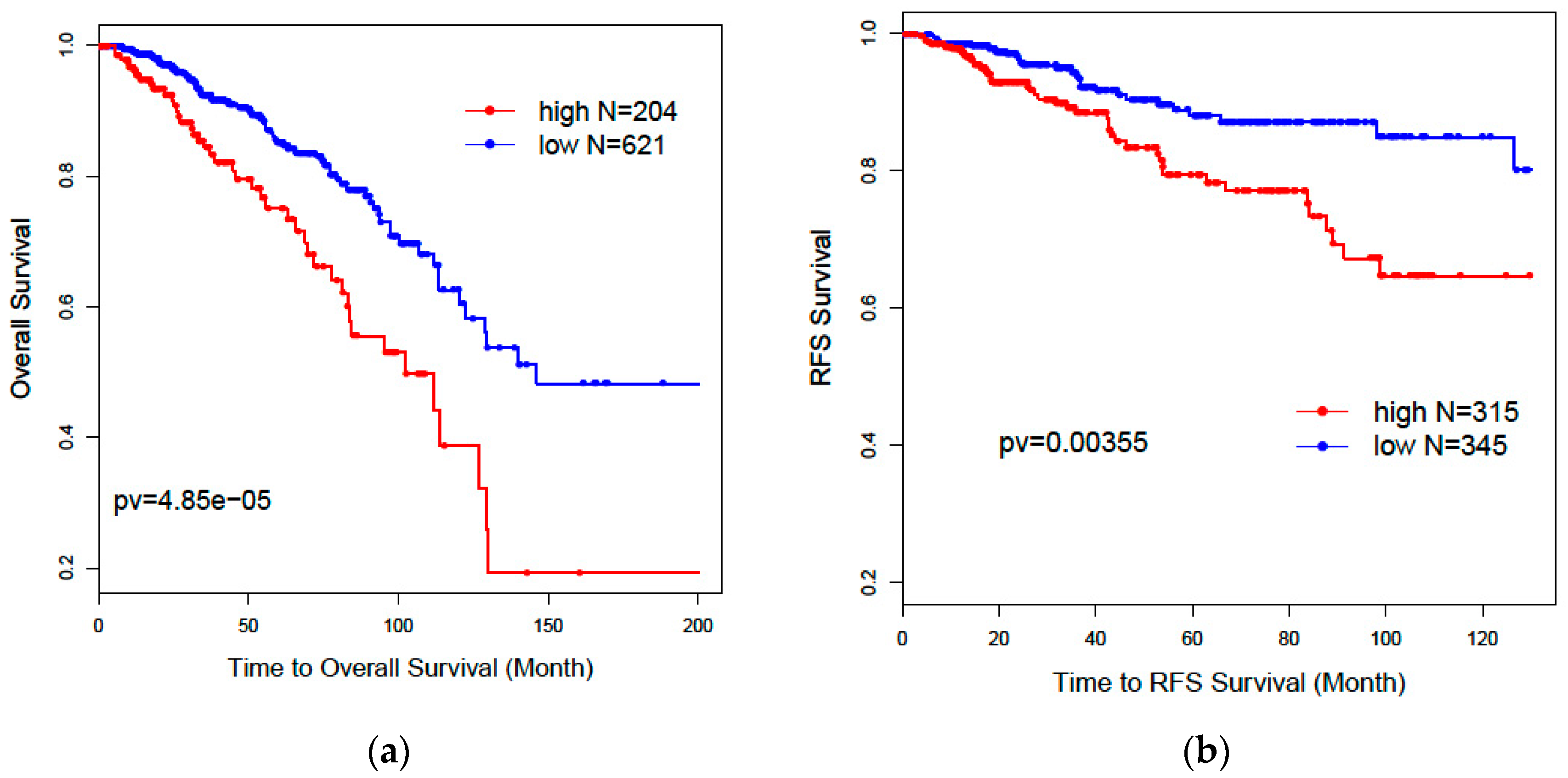
3.3. Gene Signature Evaluations Based on Clinical Data
3.4. Application of Supervised Feature Selection Strategy on Independent TCGA RNASeq Set
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Hobday, T.J.; Perez, E.A. Molecularly targeted therapies for breast cancer. Cancer Control 2005, 12, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA A Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Neo, S.-Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Zhao, X.; Rødland, E.A.; Tibshirani, R.; Plevritis, S. Molecular subtyping for clinically defined breast cancer subgroups. Breast Cancer Res. 2015, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Vallejos, C.S.; Gómez, H.L.; Cruz, W.R.; Pinto, J.A.; Dyer, R.R.; Velarde, R.; Suazo, J.F.; Neciosup, S.P.; León, M.; Miguel, A. Breast cancer classification according to immunohistochemistry markers: Subtypes and association with clinicopathologic variables in a peruvian hospital database. Clin. Breast Cancer 2010, 10, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Baehner, F.L.; Reis-Filho, J.S. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: A retrospective of the last decade. J. Pathol. 2010, 220, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, A.; Zou, D.; Xu, X.; Xia, L.; Yu, J.; Bajic, V.B.; Zhang, Z. LncRNAWiki: Harnessing community knowledge in collaborative curation of human long non-coding RNAs. Nucleic Acids Res. 2014, 43, D187–D192. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 2013, 26, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Wahlestedt, C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov. 2013, 12, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.S.; Hu, M.; Sinha, G.; Walker, N.D.; Sherman, L.S.; Gallagher, A.; Rameshwar, P. Non-coding RNA as mediators in microenvironment–breast cancer cell communication. Cancer Lett. 2016, 380, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Araya, C.L.; Cenik, C.; Reuter, J.A.; Kiss, G.; Pande, V.S.; Snyder, M.P.; Greenleaf, W.J. Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations. Nat. Genet. 2016, 48, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chinnaiyan, A.M. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Leone, S.; Santoro, R. Challenges in the analysis of long noncoding RNA functionality. FEBS Lett. 2016, 590, 2342–2353. [Google Scholar] [CrossRef] [PubMed]
- Signal, B.; Gloss, B.S.; Dinger, M.E. Computational approaches for functional prediction and characterisation of long noncoding RNAs. Trends Genet. 2016, 32, 620–637. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Zhu, J.; Rosset, S.; Tibshirani, R.; Hastie, T.J. 1-norm support vector machines. In Advances in Neural Information Processing Systems, Proceedings of the16th International Conference on Neural Information Processing Systems, Whistler, BC, Canada, 9–11 December 2003; Neural Information Processing Systems Foundation Inc.: San Diego, CA, USA, 2003. [Google Scholar]
- Díaz-Uriarte, R.; De Andres, S.A. Gene selection and classification of microarray data using random forest. BMC Bioinformatics 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Li, Y.; Wang, Z.; Zhou, S.; Sun, H.; Yu, G. The Evolution and Expression Pattern of Human Overlapping lncRNA and Protein-coding Gene Pairs. Sci. Rep. 2017, 7, 42775. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Vucic, E.A.; Enfield, K.S.; Stewart, G.L.; Lonergan, K.M.; Kennett, J.Y.; Becker-Santos, D.D.; MacAulay, C.E.; Lam, S.; Brown, C.J. Human cancer long non-coding RNA transcriptomes. PLoS ONE 2011, 6, e25915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.Y.; Ghoshal, T.; Wilkins, D.; Chen, Y.; Zhou, Y. Novel Gene Selection Methods For Breast Cancer Intrinsic Subtypes From Two Large Cohorts Study. In Proceedings of the 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Kansas City, MO, USA, 13–16 November 2017. [Google Scholar]
- Maaten, L.v.d.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Bair, E.; Hastie, T.; Paul, D.; Tibshirani, R. Prediction by supervised principal components. J. Am. Stat. Assoc. 2006, 101, 119–137. [Google Scholar] [CrossRef]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Denoeud, F.; Frankish, A.; Reymond, A.; Chen, C.-K.; Chrast, J.; Lagarde, J.; Gilbert, J.G.; Storey, R.; Swarbreck, D. GENCODE: Producing a reference annotation for ENCODE. Genome Biol. 2006, 7, S4. [Google Scholar] [CrossRef] [PubMed]
- Bair, E.; Tibshirani, R. Semi-supervised methods for predicting patient survival from gene expression papers. PLoS Biol. 2004, 2, 5011–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraley, C.; Raftery, A.E. MCLUST: Software for model-based cluster analysis. J. Classif. 1999, 16, 297–306. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Bastien, R.R.; Rodríguez-Lescure, Á.; Ebbert, M.T.; Prat, A.; Munárriz, B.; Rowe, L.; Miller, P.; Ruiz-Borrego, M.; Anderson, D.; Lyons, B. PAM50 breast cancer subtyping by RT-qPCR and concordance with standard clinical molecular markers. BMC Med.Genom. 2012, 5, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Oh, D.S.; Wessels, L.; Weigelt, B.; Nuyten, D.S.; Nobel, A.B.; Van’t Veer, L.J.; Perou, C.M. Concordance among gene-expression–based predictors for breast cancer. N. Engl. J. Med. 2006, 355, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Zhang, J.; Li, W.; Li, Z.; Hu, S.; Suo, J.; Ying, X. A Tumor-specific prognostic long non-coding RNA signature in gastric cancer. Med. Sci. Monit. 2016, 22, 3647–3657. [Google Scholar] [CrossRef] [PubMed]
- Pilato, B.; De Summa, S.; Danza, K.; Lacalamita, R.; Lambo, R.; Sambiasi, D.; Paradiso, A.; Tommasi, S. Genetic risk transmission in a family affected by familial breast cancer. J. Hum. Genet. 2014, 59, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Silina, K.; Zayakin, P.; Kalnina, Z.; Ivanova, L.; Meistere, I.; Endzelinš, E.; Abols, A.; Stengrevics, A.; Leja, M.; Ducena, K. Sperm-associated antigens as targets for cancer immunotherapy: Expression pattern and humoral immune response in cancer patients. J. Immunother. 2011, 34, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.-C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Pang, D.; Kong, D.; Chen, Q.; Xu, S.; Ping, Y. Oncogenic long noncoding RNA landscape in breast cancer. Mol. Cancer 2017, 16, 129. [Google Scholar] [CrossRef]
- Vu, T.N.; Pramana, S.; Calza, S.; Suo, C.; Lee, D.; Pawitan, Y. Comprehensive landscape of subtype-specific coding and non-coding RNA transcripts in breast cancer. Oncotarget 2016, 7, 68851–68863. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Xie, B.; Ma, Z.; Yu, W.; Wang, W.; Xu, D.; Yan, X.; Chen, B.; Yu, L.; Li, J. Identification of novel long non-coding RNAs in triple-negative breast cancer. Oncotarget 2015, 6, 21730–21739. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Type | # Of Genes before Selection | # Of Selected Genes from RNAseq | # Of PAM50 Genes from Microarray | Integrative Classification Accuracy (%) 1 |
|---|---|---|---|---|
| PCGs | 19797 | 100 | 22 | 95.5: [95.1, 95.9] |
| lncRNAs | 40701 | 85 | 21 | 95.3: [94.8, 95.8] |
| all | 60498 | 106 | 19 | 95.8: [95.4, 96.2] |
| Gene Type | # Of Genes Selected in Iteration 1 | # Of Genes Selected in Iteration 2 | Classification Accuracy (%) 1 |
|---|---|---|---|
| coding | 417 | 50 | 87.6: [87.2, 88.0] |
| non-coding | 466 | 29 | 87.8: [87.6, 88.0] |
| all | 530 | 36 | 88.5: [88.1, 88.9] |
| Gene Type | # Of Genes | Overall Survival | Recurrence-Free Survival | ||
|---|---|---|---|---|---|
| R2 | p-Value | R2 | p-Value | ||
| coding | 50 | 0.031 | 0.000000485 | 0.018 | 0.000588 |
| non-coding | 29 | 0.023 | 0.0000104 | 0.017 | 0.000938 |
| all | 36 | 0.023 | 0.00000852 | 0.025 | 0.0000483 |
| Hazards Ratio (95% Confidence Interval) | p-Value | |
|---|---|---|
| Risk Score | 2.3: [1.2, 4.5] | 0.01 * |
| Age at dignosis | 1.0: [1.0, 1.0] | 0.0041 * |
| Race | ||
| White | Reference | |
| Black | 1.5: [0.7, 3.0] | 0.31 |
| Asian | 0.5: [0.1, 3.9] | 0.51 |
| Treatment | ||
| Untreated or other | Reference | |
| Chemotherapy | 0.6: [0.3, 1.3] | 0.20 |
| Radiation therapy | 0.5: [0.2, 1.0] | 0.06 |
| Hormone therapy | 0.4: [0.1, 1.2] | 0.09 |
| Radiation & chemotherapy | 0.3: [0.1, 0.6] | 0.0027 * |
| Radiation & hormone | 0.3: [0.1, 0.8] | 0.02 * |
| Tumor stage | ||
| T1 | Reference | |
| T2 | 1.4: [0.7, 2.8] | 0.33 |
| T3 | 1.4: [0.6, 3.6] | 0.45 |
| T4 | 1.7: [0.6, 5.6] | 0.39 |
| Histology Type | ||
| Infiltrating Ductal Carcinoma | Reference | |
| Infiltrating Lobular Carcinoma | 1.2: [0.5, 3.2] | 0.70 |
| Mucinous Carcinoma | 2.6: [0.3, 22.3] | 0.83 |
| Mixed Histology | 0.9: [0.3, 2.9] | 0.37 |
| Gene_Name | Gene_Type | Chrom (Start–End Position) | HR (95% CI) | p-Value |
|---|---|---|---|---|
| DDX51 | PCGs | chr12:132136594-132144335 | 0.90: [0.84, 0.98] | 0.009 * |
| SPAG17 | PCGs | chr1: 117953861-118185223 | 0.94: [0.89, 0.99] | 0.027 * |
| NUMA1 | PCGs | chr11: 72002864-72080693 | 0.91: [0.87, 0.96] | 0.0003.5 * |
| CTD-2616J11.9 | lncRNAs | chr19: 51345169-51353293 | 0.91: [0.87, 0.96] | 0.001 * |
| RP1-140K8.1 | lncRNAs | chr6: 3893126-3894292 | 1.06: [1.00, 1.13] | 0.033 * |
| RP11-546K22.1 | lncRNAs | chr8: 51961458-52022974 | 0.94: [0.89, 0.99] | 0.043 * |
| AC000095.9 | lncRNAs | chr22: 19018043-19018916 | 0.91: [0.85, 0.98] | 0.011 * |
| SCGB1D5P | lncRNAs | chr4: 165517255-165517501 | 1.03: [1.00, 1.08] | 0.067 |
| Predicted Subtypes | ER-/PR-/HER2- | ER-/PR-/HER2+ | ER+/PR+/HER2 |
|---|---|---|---|
| Basal | 23 (92%) | 0 | 2 (2.1%) |
| Her2 | 1 (4%) | 4 (80%) | 2 (2.1%) |
| Luminal A | 1 (4%) | 1 (20%) | 76 (80.9%) |
| Luminal B | 0 | 0 | 14 (14.9%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Wang, J.; Ghoshal, T.; Wilkins, D.; Mo, Y.-Y.; Chen, Y.; Zhou, Y. lncRNA Gene Signatures for Prediction of Breast Cancer Intrinsic Subtypes and Prognosis. Genes 2018, 9, 65. https://doi.org/10.3390/genes9020065
Zhang S, Wang J, Ghoshal T, Wilkins D, Mo Y-Y, Chen Y, Zhou Y. lncRNA Gene Signatures for Prediction of Breast Cancer Intrinsic Subtypes and Prognosis. Genes. 2018; 9(2):65. https://doi.org/10.3390/genes9020065
Chicago/Turabian StyleZhang, Silu, Junqing Wang, Torumoy Ghoshal, Dawn Wilkins, Yin-Yuan Mo, Yixin Chen, and Yunyun Zhou. 2018. "lncRNA Gene Signatures for Prediction of Breast Cancer Intrinsic Subtypes and Prognosis" Genes 9, no. 2: 65. https://doi.org/10.3390/genes9020065
APA StyleZhang, S., Wang, J., Ghoshal, T., Wilkins, D., Mo, Y. -Y., Chen, Y., & Zhou, Y. (2018). lncRNA Gene Signatures for Prediction of Breast Cancer Intrinsic Subtypes and Prognosis. Genes, 9(2), 65. https://doi.org/10.3390/genes9020065