RhoB: Team Oncogene or Team Tumor Suppressor?


Abstract
:1. Introduction
2. Structure and Localization of RhoB
3. RhoB Regulation and Expression
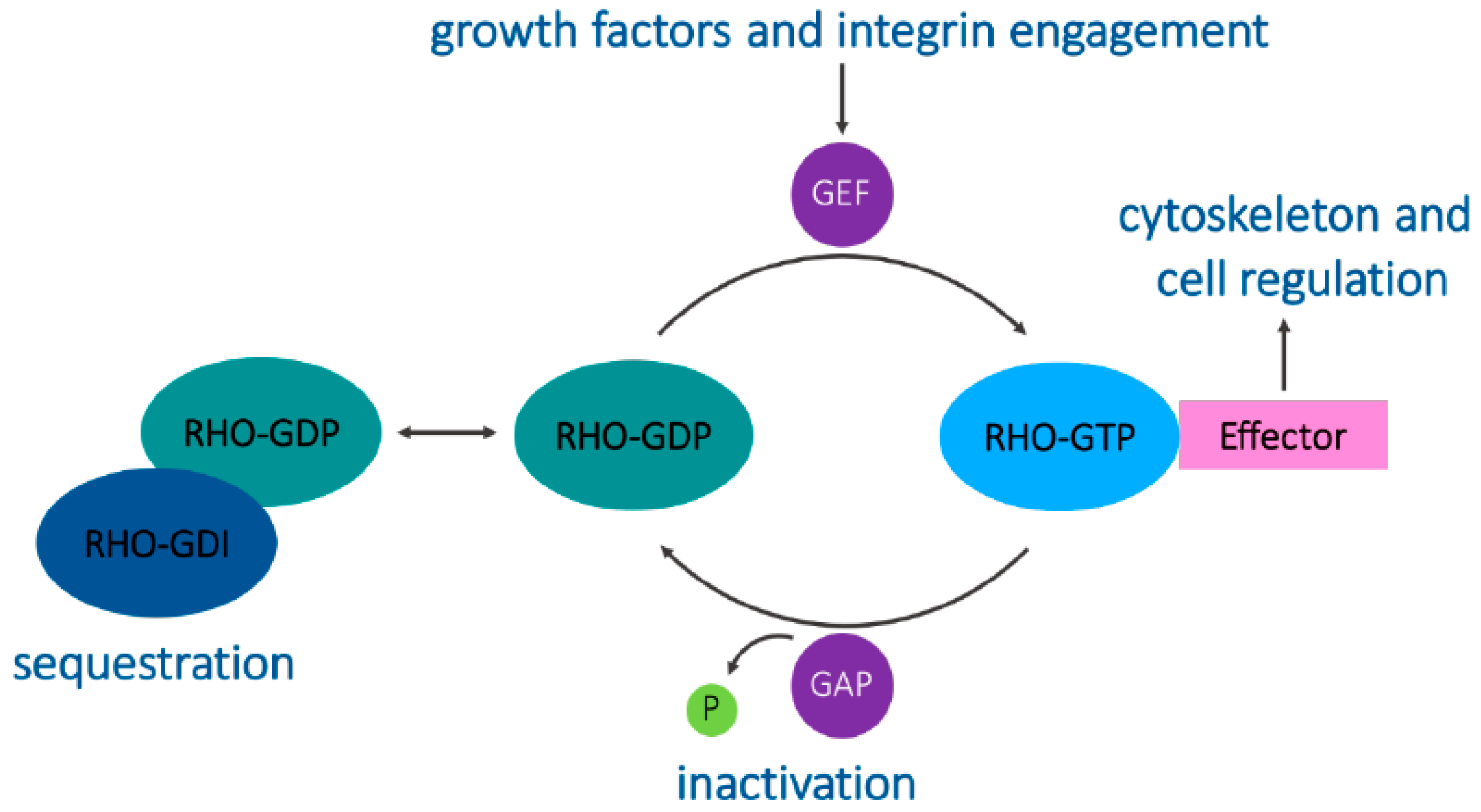
4. Signaling and Trafficking by RhoB
5. RhoB in Cancer
6. Oncogenic Role of RhoB
6.1. Glioblastoma
6.2. Breast Cancer
6.3. Lung Adenocarcinoma
6.4. Other Cancers
7. Tumor Suppressive Role
7.1. Lung Cancer
7.2. Skin Cancer
7.3. Brain Cancer
7.4. Ovarian Cancer
7.5. Other Cancers
8. Angiogenic Role
9. RhoB in FTI Anticancer Response
10. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, S.I.J.; Collard, J.G. Rho GTPases: Functions and association with cancer. Clin. Exp. Metastasis 2007, 24, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, D.T.; Brenner, J.C.; Merajver, S.D. Rho Proteins in Cancer. In The Rho GTPases in Cancer; Springer: New York, NY, USA, 2010; pp. 29–42, 135–153. ISBN 978-14-4-191110-0. [Google Scholar]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 1248, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Bagrodia, S.; Cerione, R.; Manor, D. A novel Cdc42Hs mutant induces cellular transformation. Curr. Biol. 1997, 7, 794–797. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Luo, Y.; Zheng, Y. A novel strategy for specifically down-regulating individual Rho GTPase activity in tumor cells. J. Biol. Chem. 2003, 278, 44617–44625. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. Rho–Gtpases and Cancer. Nat. Rev. Cancer 2002, 2, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C. Actin’ up: RhoB in cancer and apoptosis. Nat. Rev. Cancer 2001, 1, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Zandvakili, I.; Lin, Y.; Morris, J.C.; Zheng, Y. Rho GTPases: Anti- or pro-neoplastic targets? Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, A.; Imai, T.; Wang, C.; Ohira, S.; Feng, Y.; Nikaido, T.; Konishi, I. Up-regulation of small GTPases, RhoA and RhoC, is associated with tumor progression in ovarian carcinoma. Lab. Investig. 2003, 83, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Pillé, J.-Y.; Denoyelle, C.; Varet, J.; Bertrand, J.-R.; Soria, J.; Opolon, P.; Lu, H.; Pritchard, L.-L.; Vannier, J.-P.; Malvy, C.; et al. Anti-RhoA and anti-RhoC siRNAs inhibit the proliferation and invasiveness of MDA-MB-231 breast cancer cells in vitro and in vivo. Mol. Ther. 2005, 11, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Van Golen, K.L.; Wu, Z.F.; Xiao, X.T.; Bao, L.W.; Merajver, S.D. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000, 60, 5832–5838. [Google Scholar] [PubMed]
- Chen, Z.; Sun, J.; Pradines, A.; Favre, G.; Adnane, J.; Sebti, S.M. Both farnesylated and geranylgeranylated RhoB inhibit malignant transformation and suppress human tumor growth in nude mice. J. Biol. Chem. 2000, 275, 17974–17978. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Ridley, A.J. The RhoB small GTPase in physiology and disease. Small GTPases 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.P.; Ridley, A.J. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp. Cell Res. 2004, 301, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wherlock, M.; Gampel, A.; Futter, C.; Mellor, H. Farnesyltransferase inhibitors disrupt EGF receptor traffic through modulation of the RhoB GTPase. J. Cell Sci. 2004, 117, 3221–3231. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Khosravi-Far, R.; Solski, P.A.; Kurzawa, H.; Lebowitz, P.F.; Der, C.J. Critical role of Rho in cell transformation by oncogenic Ras. Oncogene 1995, 10, 2289–2296. [Google Scholar] [PubMed]
- Rodriguez, P.L.; Sahay, S.; Olabisi, O.O.; Whitehead, I.P. ROCK I-mediated activation of NF-κB by RhoB. Cell Signal. 2007, 19, 2361–2369. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.X.; Du, W.; Liu, J.P.; Jessell, T.M.; Prendergast, G.C. RhoB alteration is necessary for apoptotic and antineoplastic responses to farnesyltransferase inhibitors. Mol. Cell. Biol. 2000, 20, 6105–6113. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, G.; Closson, V.; Linares-Cruz, G.; Lerebours, F.; Honore, N.; Tavitian, A.; Olofsson, B. Regulation of Ras-related RhoB protein expression during the cell cycle. Oncogene 1995, 10, 1935–1945. [Google Scholar] [PubMed]
- Fritz, G.; Kaina, B.; Aktories, K. The Ras-related small GTP-binding protein RhoB is immediate-early inducible by DNA damaging treatments. J. Biol. Chem. 1995, 270, 25172–25177. [Google Scholar] [CrossRef] [PubMed]
- Jähner, D.; Hunter, T. The ras-related gene rhoB is an immediate-early gene inducible by v-Fps, epidermal growth factor, and platelet-derived growth factor in rat fibroblasts. Mol. Cell. Biol. 1991, 11, 3682–3690. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-X.; Li, Z.-B.; Diao, F.; Cao, D.-M.; Fu, C.-C.; Lu, J. Up-regulation of RhoB by glucocorticoids and its effects on the cell proliferation and NF-kappaB transcriptional activity. J. Steroid Biochem. Mol. Biol. 2006, 101, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.E.; Datta, P.K.; Moses, H.L. RhoB is stabilized by transforming growth factor beta and antagonizes transcriptional activation. J. Biol. Chem. 1998, 273, 9921–9926. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Kaina, B. Ras-related GTPase RhoB forces alkylation-induced apoptotic cell death. Biochem. Biophys. Res. Commun. 2000, 268, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Kaina, B. rhoB encoding a UV-inducible ras-related small GTP-binding protein is regulated by GTPases of the rho family and independent of JNK, ERK, and p38 MAP kinase. J. Biol. Chem. 1997, 272, 30637–30644. [Google Scholar] [CrossRef] [PubMed]
- Gampel, A.; Parker, P.J.; Mellor, H. Regulation of epidermal growth factor receptor traffic by the small GTPase RhoB. Curr. Biol. 1999, 9, 955–958. [Google Scholar] [CrossRef]
- Jiang, K.; Sun, J.; Cheng, J.; Djeu, J.Y.; Wei, S.; Sebti, S. Akt mediates Ras downregulation of RhoB, a suppressor of transformation, invasion, and metastasis. Mol. Cell. Biol. 2004, 24, 5565–5576. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Tovar, D.; He, B.; Nieto-Acosta, J.; Marty-Detraves, C.; Clanet, C.; Pradines, A.; Jablons, D.; Favre, G.; Mazières, J.; et al. Epigenetic regulation of RhoB loss of expression in lung cancer. BMC Cancer 2007, 7, 220. [Google Scholar] [CrossRef] [PubMed]
- Delarue, F.L.; Adnane, J.; Joshi, B.; Blaskovich, M.A.; Wang, D.A.; Hawker, J.; Bizouarn, F.; Ohkanda, J.; Zhu, K.; Hamilton, A.D.; et al. Farnesyltransferase and geranylgeranyltransferase I inhibitors upregulate RhoB expression by HDAC1 dissociation, HAT association and histone acetylation of the RhoB promoter. Oncogene 2007, 26, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Choi, J.H.; Won, M.; Kang, C.M.; Gyun, M.R.; Park, H.M.; Kim, C.H.; Chung, K.S. The activation of p38 MAPK primarily contributes to UV-induced RhoB expression by recruiting the c-Jun and p300 to the distal CCAAT box of the RhoB promoter. Biochem. Biophys. Res. Commun. 2011, 409, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Im, J.Y.; Han, G.; Lee, W.J.; Won, K.J.; Chung, K.S.; Lee, K.; Ban, H.S.; Song, K.; Won, M. P300 cooperates with c-Jun and PARP-1 at the p300 binding site to activate RhoB transcription in NSC126188-mediated apoptosis. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Marlow, L.A.; Bok, I.; Smallridge, R.C.; Copland, J.A. RhoB upregulation leads to either apoptosis or cytostasis through differential target selection. Endocr. Relat. Cancer 2015, 22, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, N.; Ren, K.; Meng, S.; Xie, Y.; Long, Q.; Chen, X.; Zhao, X. Expression loss and revivification of RhoB gene in ovary carcinoma carcinogenesis and development. PLoS ONE 2013, 8, e78417. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Fukui, T.; Taniguchi, T.; Yokoyama, T.; Kondo, M.; Nagasaka, T.; Goto, Y.; Gao, W.; Ueda, Y.; Yokoi, K.; et al. RhoB is frequently downregulated in non-small-cell lung cancer and resides in the 2p24 homozygous deletion region of a lung cancer cell line. Int. J. Cancer 2007, 120, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yan-Neale, Y.; Fischer, D.; Zeremski, M.; Cai, R.; Zhu, J.; Asselbergs, F.; Hampton, G.; Cohen, D. Histone deacetylase 1 represses the small GTPase RhoB expression in human nonsmall lung carcinoma cell line. Oncogene 2003, 22, 6204–6213. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.; Mellor, H. Regulation of endocytic traffic by Rho family GTPases. Trends Cell Biol. 2000, 10, 85–88. [Google Scholar] [CrossRef]
- Mellor, H.; Flynn, P.; Nobes, C.D.; Hall, A.; Parker, P.J. PRK1 is targeted to endosomes by the small GTPase, RhoB. J. Biol. Chem. 1998, 273, 4811–4814. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, E.; Cans, C.; Fincham, V.J.; Brunton, V.G.; Mellor, H.; Prendergast, G.C.; Norman, J.C.; Superti-Furga, G.; Frame, M.C. RhoB and actin polymerization coordinate Src activation with endosome-mediated delivery to the membrane. Dev. Cell 2004, 7, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Duhadaway, J.B.; Prendergast, G.C.; Laury-Kleintop, L.D. RhoB regulates PDGFR-beta trafficking and signaling in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Adini, I.; Rabinovitz, I.; Sun, J.F.; Prendergast, G.C.; Benjamin, L.E. RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev. 2003, 17, 2721–2732. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Lebowitz, P.F.; Prendergast, G.C. Cell growth inhibition by farnesyltransferase inhibitors is mediated by gain of geranylgeranylated RhoB. Mol. Cell. Biol. 1999, 19, 1831–1840. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Prendergast, G.C. Geranylgeranylated RhoB mediates suppression of human tumor cell growth by farnesyltransferase inhibitors. Cancer Res. 1999, 59, 5492–5496. [Google Scholar] [PubMed]
- Liu, A.X.; Cerniglia, G.J.; Bernhard, E.J.; Prendergast, G.C. RhoB is required to mediate apoptosis in neoplastically transformed cells after DNA damage. Proc. Natl. Acad. Sci. USA 2001, 98, 6192–6197. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.X.; Prendergast, G.C. Geranylgeranylated RhoB is sufficient to mediate tissue-specific suppression of Akt kinase activity by farnesyltransferase inhibitors. FEBS Lett. 2000, 481, 205–208. [Google Scholar] [CrossRef]
- Liu, A.X.; Rane, N.; Liu, J.P.; Prendergast, G.C. RhoB is dispensable for mouse development, but it modifies susceptibility to tumor formation as well as cell adhesion and growth factor signaling in transformed cells. Mol. Cell. Biol. 2001, 21, 6906–6912. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Antonia, T.; Daste, G.; Muro-Cacho, C.; Berchery, D.; Tillement, V.; Pradines, A.; Sebti, S.; Favre, G. Loss of RhoB Expression in Human Lung Cancer Progression. Clin. Cancer Res. 2004, 10, 2742–2750. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H. Epidemiology of Brain Tumors. In Cancer Epidemiology: Modifiable Factors; Verma, M., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 323–342. ISBN 978-1-60327-492-0. [Google Scholar]
- Iwadate, Y.; Fukuda, K.; Matsutani, T.; Saeki, N. Intrinsic protective mechanisms of the neuron-glia network against glioma invasion. J. Clin. Neurosci. 2016, 26, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Gong, Y.; Cheng, Z.; Loganathan, S.; Kao, C.; Sarkaria, J.N.; Abel, T.W.; Wang, J. Critical functions of RhoB in support of glioblastoma tumorigenesis. Neuro Oncol. 2015, 17, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Monferran, S.; Delmas, C.; Lajoie-Mazenc, I.; Favre, G.; Toulas, C.; Cohen-Jonathan-Moyal, E. Activation of RhoB by hypoxia controls hypoxia-inducible factor-1α stabilization through glycogen synthase kinase-3 in U87 glioblastoma cells. Cancer Res. 2006, 66, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M. Implications of Hypoxia in Breast Cancer Metastasis to Bone. Int. J. Mol. Sci. 2016, 17, 1669. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.A.; Godet, I.; Ye, I.C.; Byun, J.; Jayatilaka, H.; Lee, S.J.; Xiang, L.; Samanta, D.; Lee, M.H.; Wu, P.-H.; et al. Hypoxia Selectively Enhances Integrin α 5 β 1 Receptor Expression in Breast Cancer to Promote Metastasis. Mol. Cancer Res. 2017, 15, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Hockel, M.; Vaupel, P. Tumor Hypoxia: Definitions and Current Clinical, Biologic, and Molecular Aspects. JNCI J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Ader, I.; Delmas, C.; Bonnet, J.; Rochaix, P.; Favre, G.; Toulas, C.; Cohen-Jonathan-Moyal, E. Inhibition of Rho pathways induces radiosensitization and oxygenation in human glioblastoma xenografts. Oncogene 2003, 22, 8861–8869. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Fritz, G.; Brachetti, C.; Bahlmann, F.; Schmidt, M.; Kaina, B. Rho GTPases in human breast tumours: Expression and mutation analyses and correlation with clinical parameters. Br. J. Cancer 2002, 87, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Médale-Giamarchi, C.; Lajoie-Mazenc, I.; Malissein, E.; Meunier, E.; Couderc, B.; Bergé, Y.; Filleron, T.; Keller, L.; Marty, C.; Lacroix-Triki, M.; et al. RhoB modifies estrogen responses in breast cancer cells by influencing expression of the estrogen receptor. Breast Cancer Res. 2013, 15, R6. [Google Scholar] [CrossRef] [PubMed]
- Kazerounian, S.; Gerald, D.; Huang, M.; Chin, Y.R.; Udayakumar, D.; Zheng, N.; O’Donnell, R.K.; Perruzzi, C.; Mangiante, L.; Pourat, J.; et al. RhoB differentially controls akt function in tumor cells and stromal endothelial cells during breast tumorigenesis. Cancer Res. 2013, 73, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Luis-Ravelo, D.; Antón, I.; Zandueta, C.; Valencia, K.; Pajares, M.J.; Agorreta, J.; Montuenga, L.; Vicent, S.; Wistuba, I.I.; De Las Rivas, J.; et al. RHOB influences lung adenocarcinoma metastasis and resistance in a host-sensitive manner. Mol. Oncol. 2014, 8, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Calvayrac, O.; Mazières, J.; Figarol, S.; Marty-Detraves, C.; Raymond-Letron, I.; Bousquet, E.; Farella, M.; Clermont-Taranchon, E.; Milia, J.; Rouquette, I.; et al. The RAS-related GTPase RHOB confers resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer via an AKT-dependent mechanism. EMBO Mol. Med. 2017, 9, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, P.J.; Infante, E.; Khwaja, A.; Ridley, A.J. Analysis of Rho GTPase expression in T-ALL identifies RhoU as a target for Notch involved in T-ALL cell migration. Oncogene 2013, 32, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Hirokawa, Y.S.; Ohashi, A.; Uchida, K.; Kami, D.; Watanabe, M.; Yokoi, T.; Shiraishi, T.; Wakusawa, S. RhoB enhances migration and MMP1 expression of prostate cancer DU145. Exp. Mol. Pathol. 2010, 88, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, N.; Hendry, B.M.; Sharpe, C.C. Rho isoforms have distinct and specific functions in the process of epithelial to mesenchymal transition in renal proximal tubular cells. Cell Signal. 2009, 21, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Liu, W.-R.; Tian, M.-X.; Jiang, X.-F.; Wang, H.; Zhou, P.-Y.; Ding, Z.-B.; Peng, Y.-F.; Dai, Z.; Qiu, S.-J.; et al. CCL24 contributes to HCC malignancy via RhoB- VEGFA-VEGFR2 angiogenesis pathway and indicates poor prognosis. Oncotarget 2017, 8, 5135–5148. [Google Scholar] [CrossRef] [PubMed]
- Ader, I.; Toulas, C.; Dalenc, F.; Delmas, C.; Bonnet, J.; Cohen-Jonathan, E.; Favre, G. RhoB controls the 24 kDa FGF-2-induced radioresistance in HeLa cells by preventing post-mitotic cell death. Oncogene 2002, 21, 5998–6006. [Google Scholar] [CrossRef] [PubMed]
- Canguilhem, B.; Pradines, A.; Baudouin, C.; Boby, C.; Lajoie-Mazenc, I.; Charveron, M.; Favre, G. RhoB protects human keratinocytes from UVB-induced apoptosis through epidermal growth factor receptor signaling. J. Biol. Chem. 2005, 280, 43257–43263. [Google Scholar] [CrossRef] [PubMed]
- Delmas, A.; Cherier, J.; Pohorecka, M.; Medale-Giamarchi, C.; Meyer, N.; Casanova, A.; Sordet, O.; Lamant, L.; Savina, A.; Pradines, A.; et al. The c-Jun/RHOB/AKT pathway confers resistance of BRAF-mutant melanoma cells to MAPK inhibitors. Oncotarget 2015, 6, 15250–15264. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.-J.; Zhang, W.; Ni, N.-N.; Wu, Q.; Wang, X.-P.; Lin, Y.-K.; Sun, J.-F. Expression of Rho GTPases family in melanoma cells and its influence on cytoskeleton and migration. Oncotarget 2017, 8, 30112–30122. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Peyret-Lacombe, A.; Canguilhem, B.; Medale-Giamarchi, C.; Mamouni, K.; Cristini, A.; Monferran, S.; Lamant, L.; Filleron, T.; Pradines, A.; et al. RhoB promotes cancer initiation by protecting keratinocytes from UVB-induced apoptosis but limits tumor aggressiveness. J. Investig. Dermatol. 2014, 134, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Forget, M.A.; Desrosiers, R.R.; Del Maestro, R.F.; Moumdjian, R.; Shedid, D.; Berthelet, F.; Béliveau, R. The expression of Rho proteins decreases with human brain tumor progression: Potential tumor markers. Clin. Exp. Metastasis 2002, 19, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.M.; Parolin, D.A.; Lorimer, I.A. Regulation of glioblastoma cell invasion by PKC iota and RhoB. Oncogene 2008, 27, 3587–3595. [Google Scholar] [CrossRef] [PubMed]
- Vishnu, P.; Colon-Otero, G.; Kennedy, G.T.; Marlow, L.A.; Kennedy, W.P.; Wu, K.J.; Santoso, J.T.; Copland, J.A. RhoB mediates antitumor synergy of combined ixabepilone and sunitinib in human ovarian serous cancer. Gynecol. Oncol. 2012, 124, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Couderc, B.; Pradines, A.; Rafii, A.; Golzio, M.; Deviers, A.; Allal, C.; Berg, D.; Penary, M.; Teissie, J.; Favre, G. In vivo restoration of RhoB expression leads to ovarian tumor regression. Cancer Gene Ther. 2008, 15, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Kamai, T.; Tsujii, T.; Arai, K.; Takagi, K.; Asami, H.; Ito, Y.; Oshima, H. Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin. Cancer Res. 2003, 9, 2632–2641. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.M.; Chung, K.S.; Choi, S.J.; Jung, Y.J.; Park, S.K.; Han, G.H.; Ha, J.S.; Song, K.B.; Choi, N.S.; Kim, H.M.; et al. RhoB induces apoptosis via direct interaction with TNFAIP1 in HeLa cells. Int. J. Cancer 2009, 125, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tang, Q.; Qiu, M.; Lang, N.; Li, M.; Zheng, Y.; Bi, F. MiR-21 targets the tumor suppressor RhoB and regulates proliferation, invasion and apoptosis in colorectal cancer cells. FEBS Lett. 2011, 585, 2998–3005. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhu, Y.; Zhang, G.; Liu, N.; Sun, L.; Liu, M.; Qiu, M.; Luo, D.; Tang, Q.; Liao, Z.; et al. A distinct role of RhoB in gastric cancer suppression. Int. J. Cancer 2011, 128, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Adnane, J.; Muro-Cacho, C.; Mathews, L.; Sebti, S.M.; Munoz-Antonia, T. Suppression of rho B expression in invasive carcinoma from head and neck cancer patients. Clin. Cancer Res. 2002, 8, 2225–2232. [Google Scholar] [PubMed]
- Chen, W.; Niu, S.; Ma, X.; Zhang, P.; Gao, Y.; Fan, Y.; Pang, H.; Gong, H.; Shen, D.; Gu, L.; et al. RhoB acts as a tumor suppressor that inhibits malignancy of clear cell renal cell carcinoma. PLoS ONE 2016, 11, e0157599. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Yin, H.; Zhang, H.; Fang, J.; Zheng, W.; Li, D.; Li, Y.; Cao, W.; Sun, C.; Liang, Y.; et al. Sp1-driven up-regulation of miR-19a decreases RHOB and promotes pancreatic cancer. Oncotarget 2015, 6, 17391–17403. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, S.; Furuya, F.; Shimura, H.; Hayashi, Y.; Takahashi, K.; Ohta, K.; Kobayashi, T.; Kitamura, K. Activation of the RhoB signaling pathway by thyroid hormone receptor beta in thyroid cancer cells. PLoS ONE 2014, 9, e116252. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Monferran, S.; Delmas, C.; Favre, G.; Bonnet, J.; Toulas, C.; Moyal, E.C.J. αvβ3/αvβ5 integrins-fak-rhob: A novel pathway for hypoxia regulation in glioblastoma. Cancer Res. 2009, 69, 3308–3316. [Google Scholar] [CrossRef] [PubMed]
- Calvayrac, O.; Pradines, A.; Raymond-Letron, I.; Rouquette, I.; Bousquet, E.; Lauwers-Cances, V.; Filleron, T.; Cadranel, J.; Beau-Faller, M.; Casanova, A.; et al. RhoB determines tumor aggressiveness in a murine EGFRL858R-induced adenocarcinoma model and is a potential prognostic biomarker for lepidic lung cancer. Clin. Cancer Res. 2014, 20, 6541–6550. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, E.; Calvayrac, O.; Mazières, J.; Lajoie-Mazenc, I.; Boubekeur, N.; Favre, G.; Pradines, A. RhoB loss induces Rac1-dependent mesenchymal cell invasion in lung cells through PP2A inhibition. Oncogene 2015, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.C.; de-Freitas-Junior, J.C.M.; Morgado-Diaz, J.A.; Ridley, A.J.; Klumb, C.E. Dual inhibition of histone deacetylases and phosphoinositide 3-kinases: Effects on Burkitt lymphoma cell growth and migration. J. Leukoc. Biol. 2016, 99, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Howe, G.A.; Addison, C.L. RhoB controls endothelial cell morphogenesis in part via negative regulation of RhoA. Vasc. Cell 2012, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Sabatel, C.; Malvaux, L.; Bovy, N.; Deroanne, C.; Lambert, V.; Gonzalez, M.L.A.; Colige, A.; Rakic, J.M.; Noël, A.; Martial, J.A.; et al. MicroRNA-21 exhibits antiangiogenic function by targeting RhoB expression in endothelial cells. PLoS ONE 2011, 6, e16979. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Ramiro, B.; García-Weber, D.; Barroso, S.; Feito, J.; Ortega, M.C.; Cernuda-Morollón, E.; Reglero-Real, N.; Fernández-Martín, L.; Durán, M.C.; Alonso, M.A.; et al. RhoB controls endothelial barrier recovery by inhibiting Rac1 trafficking to the cell border. J. Cell Biol. 2016, 213, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Pronk, M.C.A.; van Bezu, J.S.M.; van Nieuw Amerongen, G.P.; van Hinsbergh, V.W.M.; Hordijk, P.L. RhoA, RhoB and RhoC differentially regulate endothelial barrier function. Small GTPases 2017, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gottesbühren, U.; Garg, R.; Riou, P.; McColl, B.; Brayson, D.; Ridley, A.J. Rnd3 induces stress fibres in endothelial cells through RhoB. Biol. Open 2013, 2, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.; Padhy, L.C.; Murray, M.; Weinberg, R.A. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature 1981, 290, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Sklar, M.D. The ras oncogenes increase the intrinsic resistance of NIH 3T3 cells to ionizing radiation. Science 1988, 239, 645–647. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.G.; Weiss, M.C.; Bakanauskas, V.J.; Kelsten, M.L.; Muschel, R.J.; Endlich, B.; Ling, C.C. Synergistic Effect of the v-myc Oncogene with H-ras on Radioresistance. Cancer Res. 1990, 50, 97–102. [Google Scholar] [PubMed]
- Lebowitz, P.F.; Davide, J.P.; Prendergast, G.C. Evidence that farnesyltransferase inhibitors suppress Ras transformation by interfering with Rho activity. Mol. Cell. Biol. 1995, 15, 6613–6622. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. Farnesyltransferase inhibitors and cancer treatment: Targeting simply ras? Biochim. Biophys. Acta Rev. Cancer 1997, 1333, F51–F71. [Google Scholar] [CrossRef]
- Lebowitz, P.F.; Casey, P.J.; Prendergast, G.C.; Thissen, J.A. Farnesyltransferase inhibitors alter the prenylation and growth- stimulating function of RhoB. J. Biol. Chem. 1997, 272, 15591–15594. [Google Scholar] [CrossRef] [PubMed]
- Kamasani, U.; Huang, M.; DuHadaway, J.B.; Prochownik, E.V.; Donover, P.S.; Prendergast, G.C. Cyclin B1 is a critical target of RhoB in the cell suicide program triggered by farnesyl transferase inhibition. Cancer Res. 2004, 64, 8389–8396. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Gibbs, R.A.; Mattingly, R.R. Working together: Farnesyl transferase inhibitors and statins block protein prenylation. Mol. Cell. Pharmacol. 2009, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.J.; Ho, L.; Ranganathan, S.; Abbruzzese, J.L.; Alpaugh, R.K.; Beard, M.; Lewis, N.L.; McLaughlin, S.; Rogatko, A.; Perez-Ruixo, J.J.; et al. Phase II and pharmacodynamic study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 2003, 21, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; Mauer, A.; Bruzek, L.; Marks, R.S.; Hillman, S.; Geyer, S.; Hanson, L.J.; Wright, J.J.; Erlichman, C.; Kaufmann, S.H.; et al. Phase II Study of the Farnesyl Transferase Inhibitor R115777 in Patients With Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2003, 21, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.D.; Hickish, T.; Ellis, P.; Houston, S.; Kelland, L.; Dowsett, M.; Salter, J.; Michiels, B.; Perez-Ruixo, J.J.; Palmer, P.; et al. Phase II study of the efficacy and tolerability of two dosing regimens of the farnesyl transferase inhibitor, R115777, in advanced breast cancer. J. Clin. Oncol. 2003, 21, 2492–2499. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Cunningham, D.; de Gramont, A.; Scheithauer, W.; Smakal, M.; Humblet, Y.; Kourteva, G.; Iveson, T.; Andre, T.; Dostalova, J.; et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J. Clin. Oncol. 2004, 22, 3950–3957. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sharma, S.; Saltz, L.B.; Kemeny, N.; Kelsen, D.P.; Ilson, D.; O’Reilly, E.; Zaknoen, S.; Baum, C.; Statkevich, P.; et al. A phase II trial of farnesyl protein transferase inhibitor SCH 66336, given by twice-daily oral administration, in patients with metastatic colorectal cancer refractory to 5-fluorouracil and irinotecan. Ann. Oncol. 2002, 13, 1067–1071. [Google Scholar] [CrossRef]
- Winquist, E.; Moore, M.J.; Chi, K.N.; Ernst, D.S.; Hirte, H.; North, S.; Powers, J.; Walsh, W.; Boucher, T.; Patton, R.; et al. A multinomial Phase II study of lonafarnib (SCH 66336) in patients with refractory urothelial cancer. Urol. Oncol. Semin. Orig. Investig. 2005, 23, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; Erlichman, C.; Davis, J.N.; Cutler, D.L.; Sloan, J.A.; Marks, R.S.; Hanson, L.J.; Svingen, P.A.; Atherton, P.; Bishop, W.R.; et al. A phase I trial of the farnesyl transferase inhibitor SCH66336: Evidence for biological and clinical activity. Cancer Res. 2000, 60, 1871–1877. [Google Scholar] [PubMed]
- Ho, C.L.; Kurman, R.J.; Dehari, R.; Wang, T.L.; Shih, I.M. Mutations of BRAF and KRAS precede the development of ovarian serous borderline tumors. Cancer Res. 2004, 64, 6915–6918. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.M.; Eisen, T.G. Farnesyl transferase inhibitor SCH66336 is cytostatic, pro-apoptotic and enhances chemosensitivity to cisplatin in melanoma cells. Int. J. Cancer 2003, 105, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Halaschek-Wiener, J.; Kloog, Y.; Wacheck, V.; Jansen, B. Farnesyl thiosalicylic acid chemosensitizes human melanoma in vivo. J. Investig. Dermatol. 2003, 120, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Liao, Y.P.; Mischel, P.S.; Iwamoto, K.S.; Cacalano, N.A.; McBride, W.H. HDJ-2 as a target for radiosensitization of glioblastoma multiforme cells by the farnesyltransferase inhibitor R115777 and the role of the p53/p21 pathway. Cancer Res. 2006, 66, 6756–6762. [Google Scholar] [CrossRef] [PubMed]
- Lebowitz, P.F.; Prendergast, G.C. Non-Ras targets of farnesyltransferase inhibitors: Focus on Rho. Oncogene 1998, 17, 1439–1445. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cancer Type | Oncogene | Tumor Suppressor |
|---|---|---|
| Bladder | [77] ‡ | |
| Brain | [52] §, [53], [85], [57] § | [73] ‡, [74] |
| Breast | [58] ‡, [59] ‡, [60] §,‡, [61] *,§ | [61] *,§ |
| Cervical | [68] | [78] ‡ |
| Colorectal | [79] ‡ | |
| Gastric | [80] ‡ | |
| Head and Neck | [81] ‡ | |
| Kidney | [66] | [82] ‡ |
| Liver | [67] *,§,‡ | |
| Lung | [62] §,‡, [63] §,‡ | [31] ‡, [37] ‡, [49] §,‡, [86] §,‡, [87] |
| Lymphoma | [64] ‡ | [88] |
| Ovarian | [36] ‡, [75] §,‡, [76] § | |
| Pancreatic | [15] §, [83] §,‡ | |
| Prostate | [65] | |
| Skin | [69], [70] §,‡, [72] §,‡ | [30] §, [48] §, [71], [72] §,‡ |
| Thyroid | [84] §,‡ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, J.A.; Gilkes, D.M. RhoB: Team Oncogene or Team Tumor Suppressor? Genes 2018, 9, 67. https://doi.org/10.3390/genes9020067
Ju JA, Gilkes DM. RhoB: Team Oncogene or Team Tumor Suppressor? Genes. 2018; 9(2):67. https://doi.org/10.3390/genes9020067
Chicago/Turabian StyleJu, Julia A., and Daniele M. Gilkes. 2018. "RhoB: Team Oncogene or Team Tumor Suppressor?" Genes 9, no. 2: 67. https://doi.org/10.3390/genes9020067
APA StyleJu, J. A., & Gilkes, D. M. (2018). RhoB: Team Oncogene or Team Tumor Suppressor? Genes, 9(2), 67. https://doi.org/10.3390/genes9020067