Exploration and Exploitation of Novel SSR Markers for Candidate Transcription Factor Genes in Lilium Species

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Plant Materials
2.2. cDNA Library Construction
2.3. Transcriptome Sequencing and Assembling
2.4. TFSSR Marker Development and In Silico Characterization
2.5. In Silico TFSSR Expression Profiling in Response to Biotic Stress
2.6. DNA Extraction from Plant Materials for Validation of TFSSR
2.7. Experimental Validation of TFSSRs
2.8. Data Analysis
3. Results
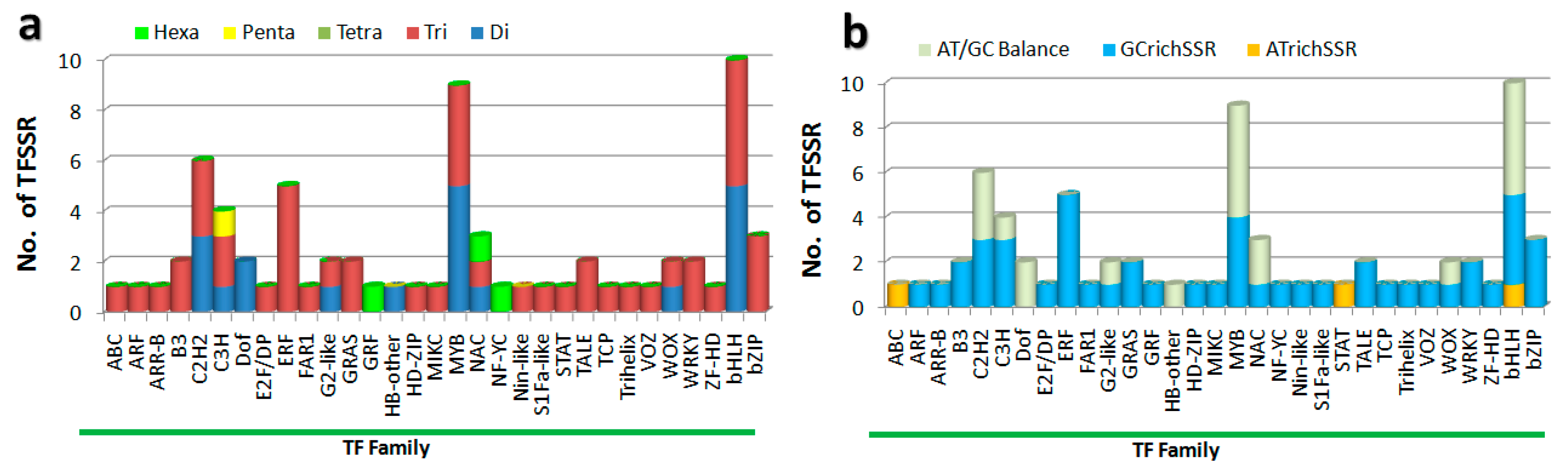
3.1. In Silico Characterization of TFSSR
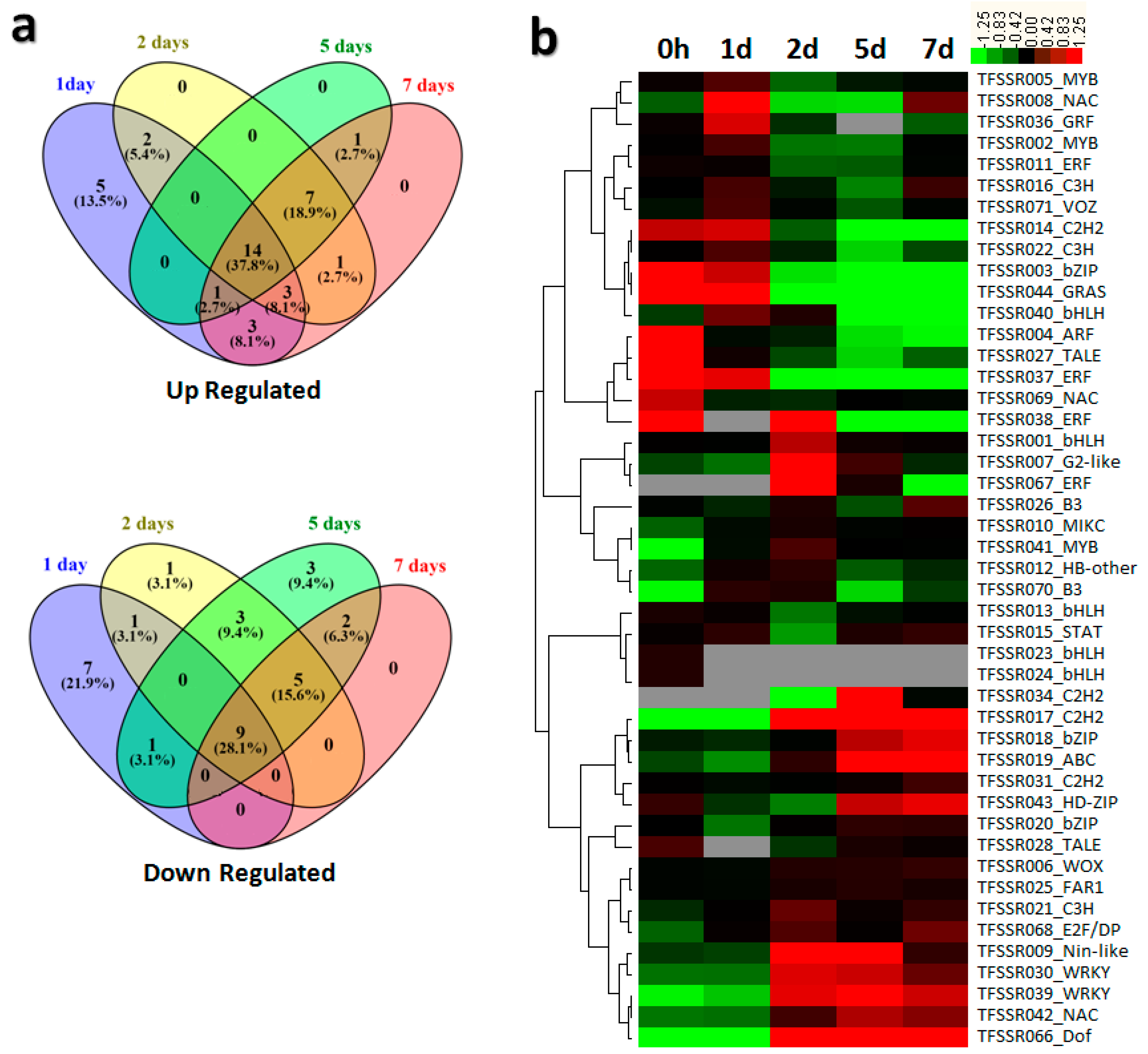
3.2. TFSSR Expression Profiling upon B. elliptica Infection
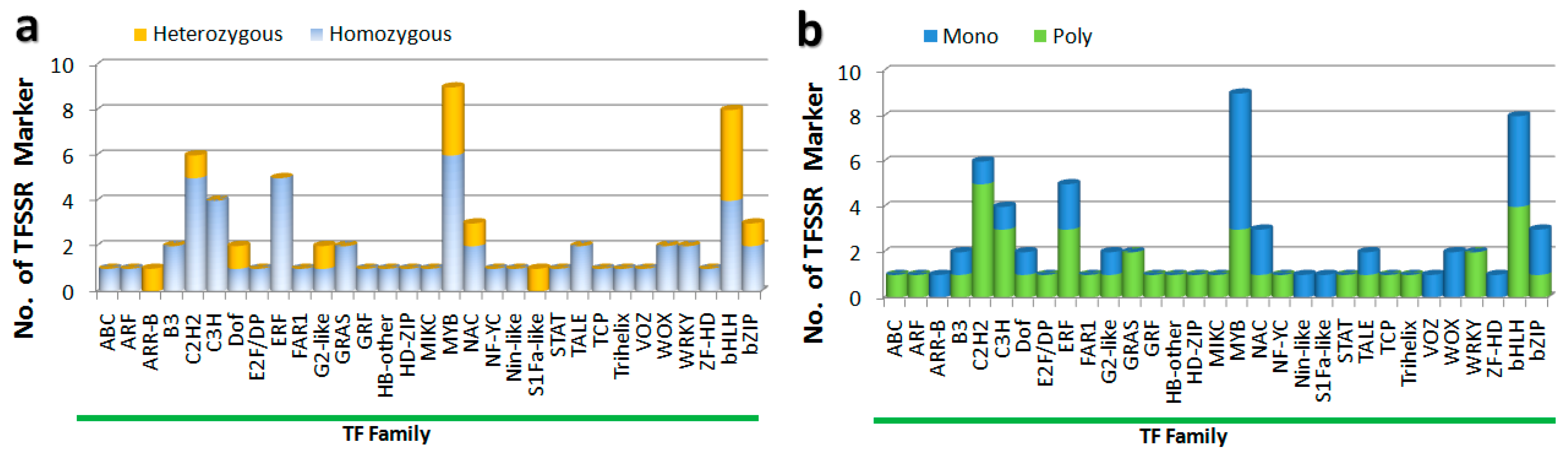
3.3. Evaluation of Amplification, Polymorphism, and Potential of TFSSRs as Markers
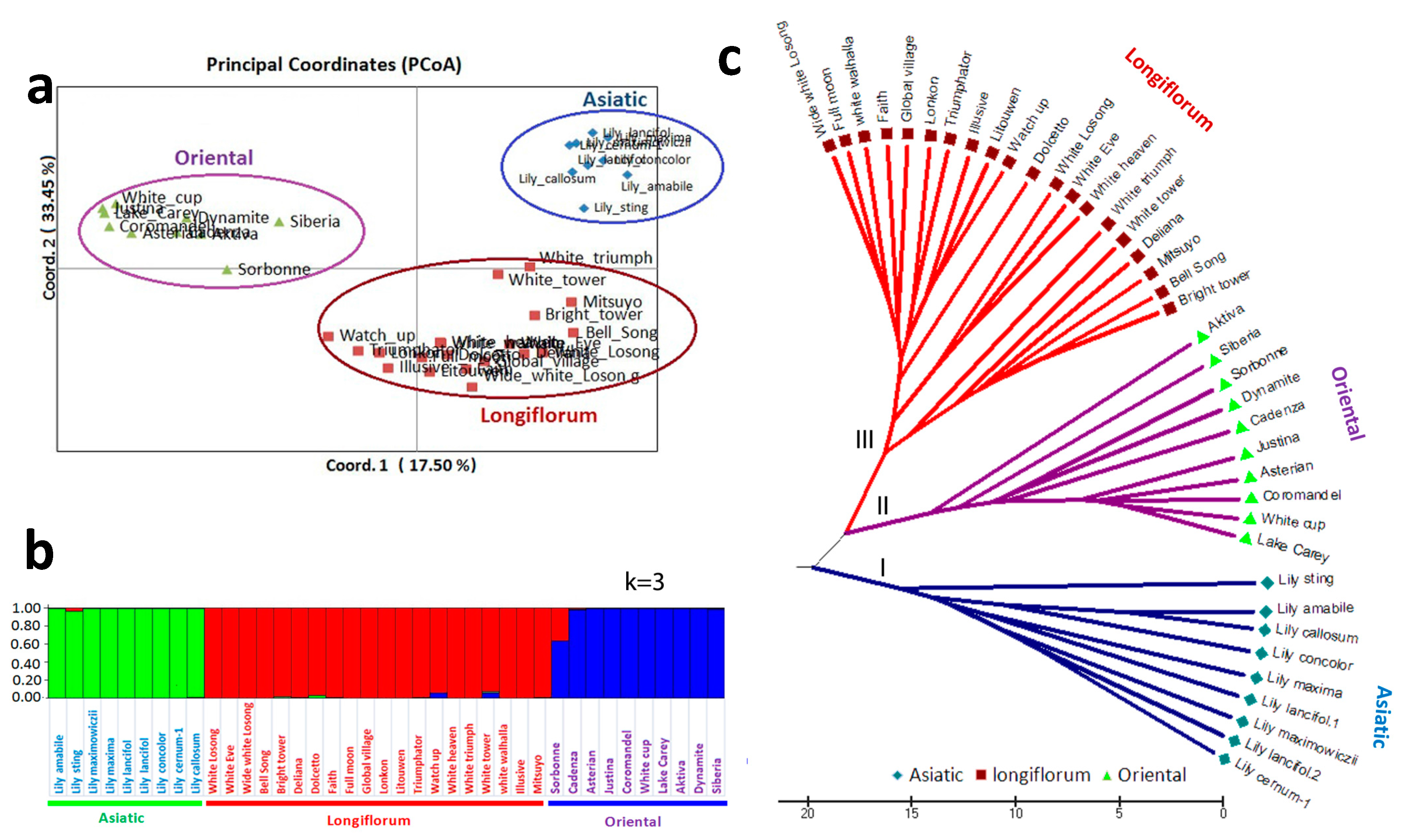
3.4. Analysis of Genetic Diversity and Population Structure Using TFSSR Markers
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Buschman, J.C.M. Globalisation - Flower - Flower Bulbs - Bulb Flowers; International Society for Horticultural Science (ISHS): Leuven, Belgium, 2004; pp. 27–33. [Google Scholar]
- Shahin, A.; van Kaauwen, M.; Esselink, D.; Bargsten, J.W.; van Tuyl, J.M.; Visser, R.G.; Arens, P. Generation and analysis of expressed sequence tags in the extreme large genomes Lilium and Tulipa. BMC Genomics 2012, 13, 640. [Google Scholar] [CrossRef] [PubMed]
- Comber, H.F. A new classification of the genus Lilium. Lily Year Book RHS 1949.
- De Jong, P. Some notes on the evolution of lilies. Lily yearbook 1974.
- Bakhshaie, M.; Khosravi, S.; Azadi, P.; Bagheri, H.; van Tuyl, J.M. Biotechnological advances in Lilium. Plant Cell Rep. 2016, 35, 1799–1826. [Google Scholar] [CrossRef] [PubMed]
- Shahin, A.; Smulders, M.J.; van Tuyl, J.M.; Arens, P.; Bakker, F.T. Using multi-locus allelic sequence data to estimate genetic divergence among four Lilium (Liliaceae) cultivars. Front. Plant Sci. 2014, 5, 567. [Google Scholar] [CrossRef] [PubMed]
- Hahm, S.-S.; Lee, K.-H.; Lee, J.-W.; Lee, H.-D.; Yu, S.-H. Control and incidence of leaf blight on lily with different cultural systems. Res. Plant Dis. 2007, 13, 152–156. [Google Scholar] [CrossRef]
- Shahin, A.; Arens, P.; Van Heusden, A.W.; Van Der Linden, G.; Van Kaauwen, M.; Khan, N.; Schouten, H.J.; De Weg, V.; Eric, W.; Visser, R.G. Genetic mapping in Lilium: Mapping of major genes and quantitative trait loci for several ornamental traits and disease resistances. Plant Breed. 2011, 130, 372–382. [Google Scholar] [CrossRef]
- Haruki, K.; Hosoki, T.; Nako, Y. Tracing the parentages of some oriental hybrid lily cultivars by PCR-RFLP analysis. J. Jpn. Soc. Hortic. Sci. 1998, 67, 352–359. [Google Scholar] [CrossRef]
- Varshney, A.; Sharma, M.P.; Adholeya, A.; Dhawan, V.; Srivastava, P. Enhanced growth of micropropagated bulblets of Lilium sp. Inoculated with arbuscular mycorrhizal fungi at different P fertility levels in an alfisol. J. Hortic. Sci. Biotechnol. 2002, 77, 258–263. [Google Scholar] [CrossRef]
- Wen, C.; Hsiao, J. Altitudinal genetic differentiation and diversity of Taiwan lily (Lilium longiflorum var. Formosanum; liliaceae) using RAPD markers and morphological characters. Int. J. Plant Sci. 2001, 162, 287–295. [Google Scholar] [CrossRef]
- Xi, M.; Sun, L.; Qiu, S.; Liu, J.; Xu, J.; Shi, J. In vitro mutagenesis and identification of mutants via ISSR in lily (Lilium longiflorum). Plant Cell Rep. 2012, 31, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Horning, M.E.; Maloney, S.C.; Webster, M.S. Isolation and characterization of variable microsatellite loci in Lilium philadelphicum (liliaceae). Mol. Ecol. Resour. 2003, 3, 412–413. [Google Scholar] [CrossRef]
- Sakazono, S.; Hiramatsu, M.; Watanabe, M.; Okubo, H. Development and characterization of microsatellite markers for Lilium longiflorum (liliaceae). Appl. Plant Sci. 2013, 1, 1300014. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-K.; Liu, C.-C.; Chiang, T.-Y.; Chen, M.-T.; Chou, C.-H.; Yeh, C.-H. Characterization of expressed sequence tags from flower buds of alpine Lilium formosanum using a subtractive cDNA library. Plant Mol. Biol. Rep. 2011, 29, 88–97. [Google Scholar] [CrossRef]
- Lee, S.-I.; Park, K.-C.; Song, Y.-S.; Son, J.-H.; Kwon, S.-J.; Na, J.-K.; Kim, J.-H.; Kim, N.-S. Development of expressed sequence tag derived-simple sequence repeats in the genus Lilium. Genes Genom. 2011, 33, 727–733. [Google Scholar] [CrossRef]
- Libault, M.; Joshi, T.; Benedito, V.A.; Xu, D.; Udvardi, M.K.; Stacey, G. Legume transcription factor genes: What makes legumes so special? Plant Physiol. 2009, 151, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Kapoor, S.; Tyagi, A.K. Transcription factors regulating the progression of monocot and dicot seed development. Bioessays 2011, 33, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-K.; Paik, H.; Choi, J.P.; Han, J.-H.; Choe, J.-K.; Hur, C.-G. Functional domain marker (fdm): An in silico demonstration in solanaceae using simple sequence repeats (SSRs). Plant Mol. Biol. Rep. 2010, 28, 352–356. [Google Scholar] [CrossRef]
- Biswas, M.K.; Peng, C.; Mohamed Hamdy, A.; Xiuxin, D. Novel polymorphic EST-based microsatellite marker isolation and characterization from Poncirus trifoliata (rutaceae). Frontiers Agric. Sci. Eng. 2015, 2, 60–65. [Google Scholar] [CrossRef]
- Biswas, M.K.; Xu, Q.; Mayer, C.; Deng, X. Genome wide characterization of short tandem repeat markers in sweet orange (Citrus sinensis). PLoS ONE 2014, 9, e104182. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.K.; Chai, L.; Qiang, X.; Deng, X. Generation, functional analysis and utility of Citrus grandis EST from a flower-derived cDNA library. Mol. Biol. Rep. 2012, 39, 7221–7235. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.K.; Chai, L.; Mayer, C.; Xu, Q.; Guo, W.; Deng, X. Exploiting bac-end sequences for the mining, characterization and utility of new short sequences repeat (SSR) markers in citrus. Mol. Biol. Rep. 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.K.; Liu, Y.; Li, C.; Sheng, O.; Mayer, C.; Yi, G. Genome-wide computational analysis of musa microsatellites: Classification, cross-taxon transferability, functional annotation, association with transposons & miRNAs, and genetic marker potential. PLoS ONE 2015, 10, e0131312. [Google Scholar]
- Cavagnaro, P.F.; Senalik, D.A.; Yang, L.; Simon, P.W.; Harkins, T.T.; Kodira, C.D.; Huang, S.; Weng, Y. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genomics 2010, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Wu, Y.; Zhang, L.; Li, X.-W.; Zhao, X.-Y.; Wang, W.-H.; Gao, Z.-S.; Xia, Y.-P. De novo assembled transcriptome analysis and SSR marker development of a mixture of six tissues from Lilium oriental hybrid ‘Sorbonne’. Plant Mol. Biol. Rep. 2015, 33, 281–293. [Google Scholar] [CrossRef]
- Yang, S.-L.; Ming, J.; Liu, C.; Mu, D.; Li, M.-Y. Data mining for simple sequence repeats marker development in expressed sequence tags from Lilium L. Acta Hortic. Sin. 2008, 7, 022. [Google Scholar]
- Yuan, S.; Ge, L.; Liu, C.; Ming, J. The development of EST-SSR markers in Lilium regale and their cross-amplification in related species. Euphytica 2013, 189, 393–419. [Google Scholar] [CrossRef]
- FastQC software. Available online: http://www.Bioinformatics.Babraham.Ac.Uk/projects/fastqc (accessed on 15 March 2017).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Lily database. Available online: http://210.110.86.160/Lidb/Lilidb_Home.html (accessed on 25 March 2017).
- Lstat: web-based micro-satellite analysis tool for family liliaceace. Available online: http://210.110.86.160/Lsat/Lsat.html (accessed on 25 March 2017).
- Rozen, S.; Skaletsky, H. Primer3 on the www for general users and for biologist programmers. Bioinformatics Methods Protocols 1999, 365–386. [Google Scholar]
- De Hoon, M.J.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. Powermarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A. Structure harvester: A website and program for visualizing structure output and implementing the evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Miao, Y.; Zhu, Z.; Guo, Q.; Zhu, Y.; Yang, X.; Sun, Y. Transcriptome analysis of differentially expressed genes provides insight into stolon formation in Tulipa edulis. Front. Plant Sci. 2016, 7, 409. [Google Scholar] [CrossRef] [PubMed]
- Kujur, A.; Bajaj, D.; Saxena, M.S.; Tripathi, S.; Upadhyaya, H.D.; Gowda, C.; Singh, S.; Jain, M.; Tyagi, A.K.; Parida, S.K. Functionally relevant microsatellite markers from chickpea transcription factor genes for efficient genotyping applications and trait association mapping. DNA Res. 2013, dst015. [Google Scholar] [CrossRef] [PubMed]
- Morgante, M.; Hanafey, M.; Powell, W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002, 30, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.J.; Shi, J.R.; Singh, S.; Fickus, E.W.; Costa, J.M.; Lewis, J.; Gill, B.S.; Ward, R.; Cregan, P.B. Development and mapping of microsatellite (SSR) markers in wheat. Theor. Appl. Genet. 2005, 110, 550–560. [Google Scholar] [CrossRef] [PubMed]
- McCouch, S.R.; Teytelman, L.; Xu, Y.; Lobos, K.B.; Clare, K.; Walton, M.; Fu, B.; Maghirang, R.; Li, Z.; Xing, Y.; et al. Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res. 2002, 9, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Wang, F.; Jiang, Q.; Ma, J.; Xiong, A.S. Identification of SSRs and differentially expressed genes in two cultivars of celery (Apium graveolens L.) by deep transcriptome sequencing. Hortic. Res. 2014, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Gaspari, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Guo, C.; Sutharzan, S.; Li, P.; Echt, C.S.; Zhang, J.; Liang, C. Genome-wide analysis of tandem repeats in plants and green algae. G3: Genes, Genomes, Genetics 2014, 4, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Udvardi, M.K.; Kakar, K.; Wandrey, M.; Montanari, O.; Murray, J.; Andriankaja, A.; Zhang, J.-Y.; Benedito, V.; Hofer, J.M.; Chueng, F. Legume transcription factors: Global regulators of plant development and response to the environment. Plant Physiol. 2007, 144, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Jiang, J.; Jia, H.; Zhao, X.-Y.; Wang, W.-H.; Gao, Q.-K.; Mao, W.-H.; Wu, Y.; Zhang, L.; Grierson, D. Selection of generally applicable ssr markers for evaluation of genetic diversity and identity in Lilium. Biochem. Syst. Ecol. 2015, 61, 278–285. [Google Scholar] [CrossRef]
- Lee, S.-I.; Nguyen, X.T.; Kim, J.-H.; Kim, N.-S. Genetic diversity and structure analyses on the natural populations of diploids and triploids of tiger lily, Lilium lancifolium Thunb., from Korea, China, and Japan. Genes Genom. 2016, 38, 467–477. [Google Scholar] [CrossRef]
- Chung, M.Y.; Chung, M.G. Large effective population sizes and high levels of gene flow between subpopulations of Lilium cernuum (liliaceae). Biochem. Syst. Ecol. 2014, 54, 354–361. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Count | % |
|---|---|---|
| No. of sequences searched | 216,768 | |
| SSR-containing sequences | 6966 | 3.21 |
| Transcription factor SSRs | 71 | 1.99 |
| Di-nucleotide repeats | 20 | 28.17 |
| Tri-nucleotide repeats | 47 | 66.20 |
| Tetra-nucleotide repeats | 0 | 0.00 |
| Penta-nucleotide repeats | 1 | 1.41 |
| Hexa-nucleotide repeats | 3 | 4.23 |
| Class I members | 11 | 15.49 |
| Class II members | 60 | 84.51 |
| GC-rich SSRs | 47 | 66.20 |
| AT-rich SSRs | 3 | 4.23 |
| AT/GC-balanced SSRs | 21 | 29.58 |
| Parameters | Di | Tri | Tetra | Penta | Hexa | Total/Average |
|---|---|---|---|---|---|---|
| Tested primer | 20 | 47 | na | 1 | 3 | 71 |
| PCR amplification | 18 | 47 | na | 1 | 3 | 69 |
| Band Specific | 9 | 28 | na | 0 | 2 | 39 |
| Scorable Primer | 11 | 31 | na | 0 | 2 | 44 |
| Polymorphic | 10 | 29 | na | 0 | 2 | 41 |
| Range of Alleles No. | 2–5 | 2–6 | na | na | 2–3 | 2–6 |
| Total No. of Alleles | 50 | 146 | na | 4 | 7 | 207 |
| No. of Homozygous | 12 | 40 | na | 1 | 2 | 55 |
| No. of Heterozygous | 6 | 7 | na | 0 | 1 | 14 |
| Homo:Hetero Ratio | 2:1 | 6:1 | na | na | 2:1 | 4:1 |
| Mean of Alleles ±SD | 2.78 ± 0.94 | 3.11 ± 1.5 | na | na | 2.33 ± 0.58 | 3.06 ± 0.755 |
| PIC±SD | 0.55 ± 0.17 | 0.64 ± 0.15 | na | 0.47 ± 0 | 0.65 ± 0.16 | 0.58 ± 0.12 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, M.K.; Nath, U.K.; Howlader, J.; Bagchi, M.; Natarajan, S.; Kayum, M.A.; Kim, H.-T.; Park, J.-I.; Kang, J.-G.; Nou, I.-S. Exploration and Exploitation of Novel SSR Markers for Candidate Transcription Factor Genes in Lilium Species. Genes 2018, 9, 97. https://doi.org/10.3390/genes9020097
Biswas MK, Nath UK, Howlader J, Bagchi M, Natarajan S, Kayum MA, Kim H-T, Park J-I, Kang J-G, Nou I-S. Exploration and Exploitation of Novel SSR Markers for Candidate Transcription Factor Genes in Lilium Species. Genes. 2018; 9(2):97. https://doi.org/10.3390/genes9020097
Chicago/Turabian StyleBiswas, Manosh Kumar, Ujjal Kumar Nath, Jewel Howlader, Mita Bagchi, Sathishkumar Natarajan, Md Abdul Kayum, Hoy-Taek Kim, Jong-In Park, Jong-Goo Kang, and Ill-Sup Nou. 2018. "Exploration and Exploitation of Novel SSR Markers for Candidate Transcription Factor Genes in Lilium Species" Genes 9, no. 2: 97. https://doi.org/10.3390/genes9020097
APA StyleBiswas, M. K., Nath, U. K., Howlader, J., Bagchi, M., Natarajan, S., Kayum, M. A., Kim, H. -T., Park, J. -I., Kang, J. -G., & Nou, I. -S. (2018). Exploration and Exploitation of Novel SSR Markers for Candidate Transcription Factor Genes in Lilium Species. Genes, 9(2), 97. https://doi.org/10.3390/genes9020097