Ultrahigh-Density Linkage Map Construction Using Low-Coverage Whole-Genome Sequencing of a Doubled Haploid Population: Case Study of Torafugu (Takifugu rubripes)

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of a Doubled-Haploid Population of Torafugu
2.2. Whole-Genome Sequencing
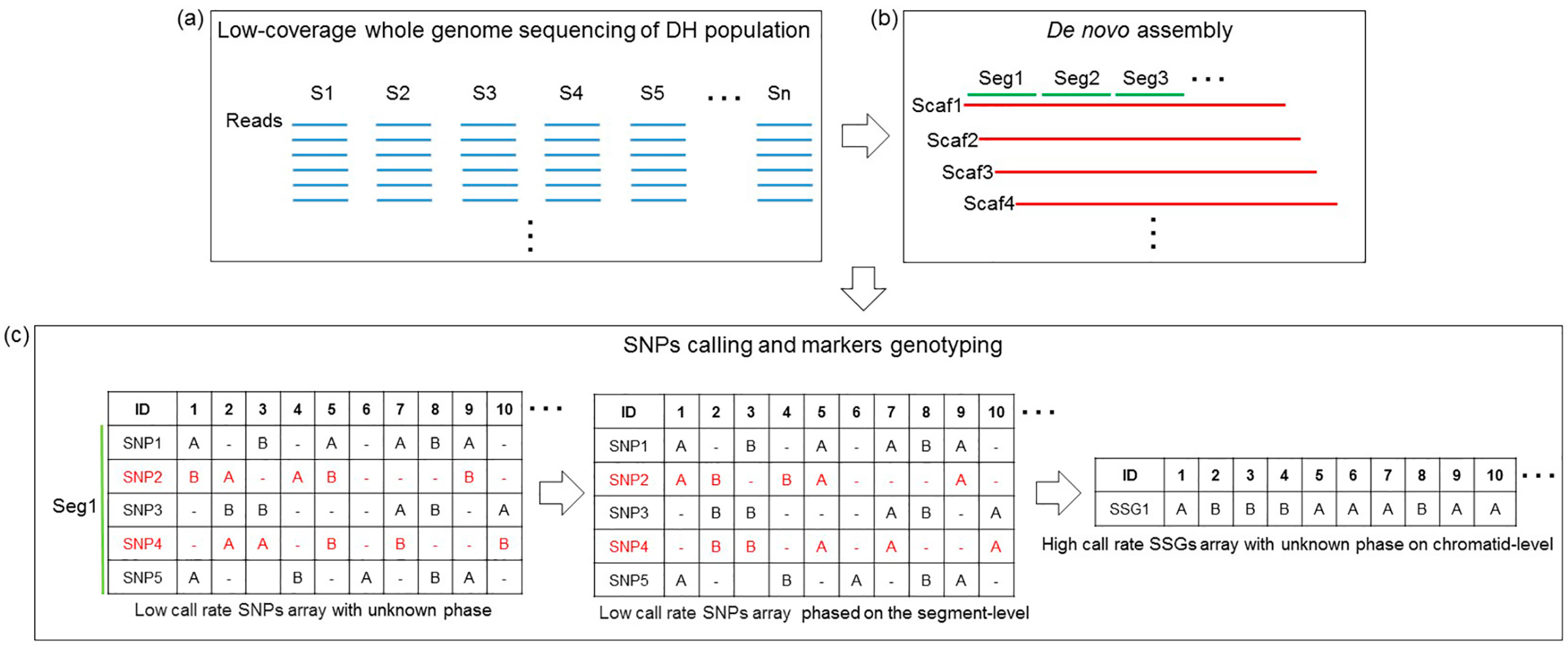
2.3. De Novo Assembly and SNP Calling
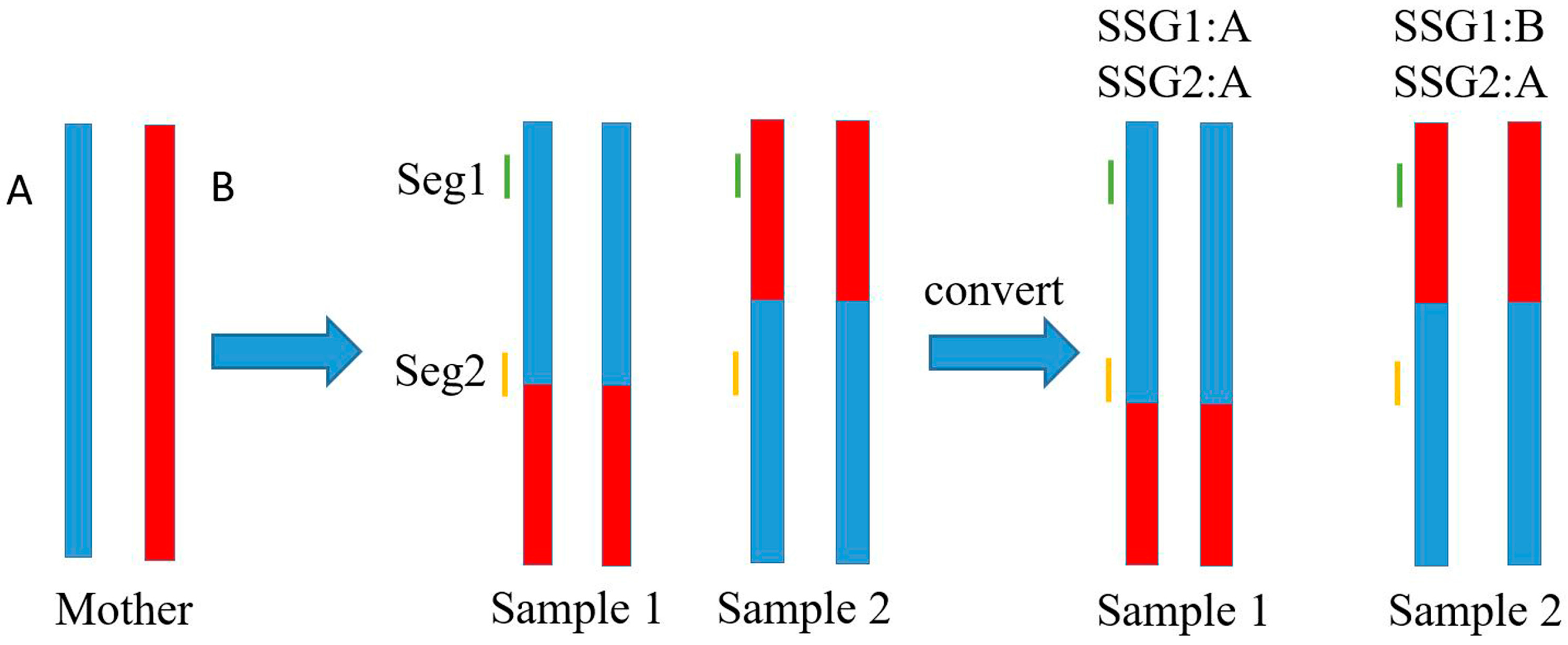
2.4. Marker Genotype Scoring
2.5. Construction of the Genetic Linkage Map
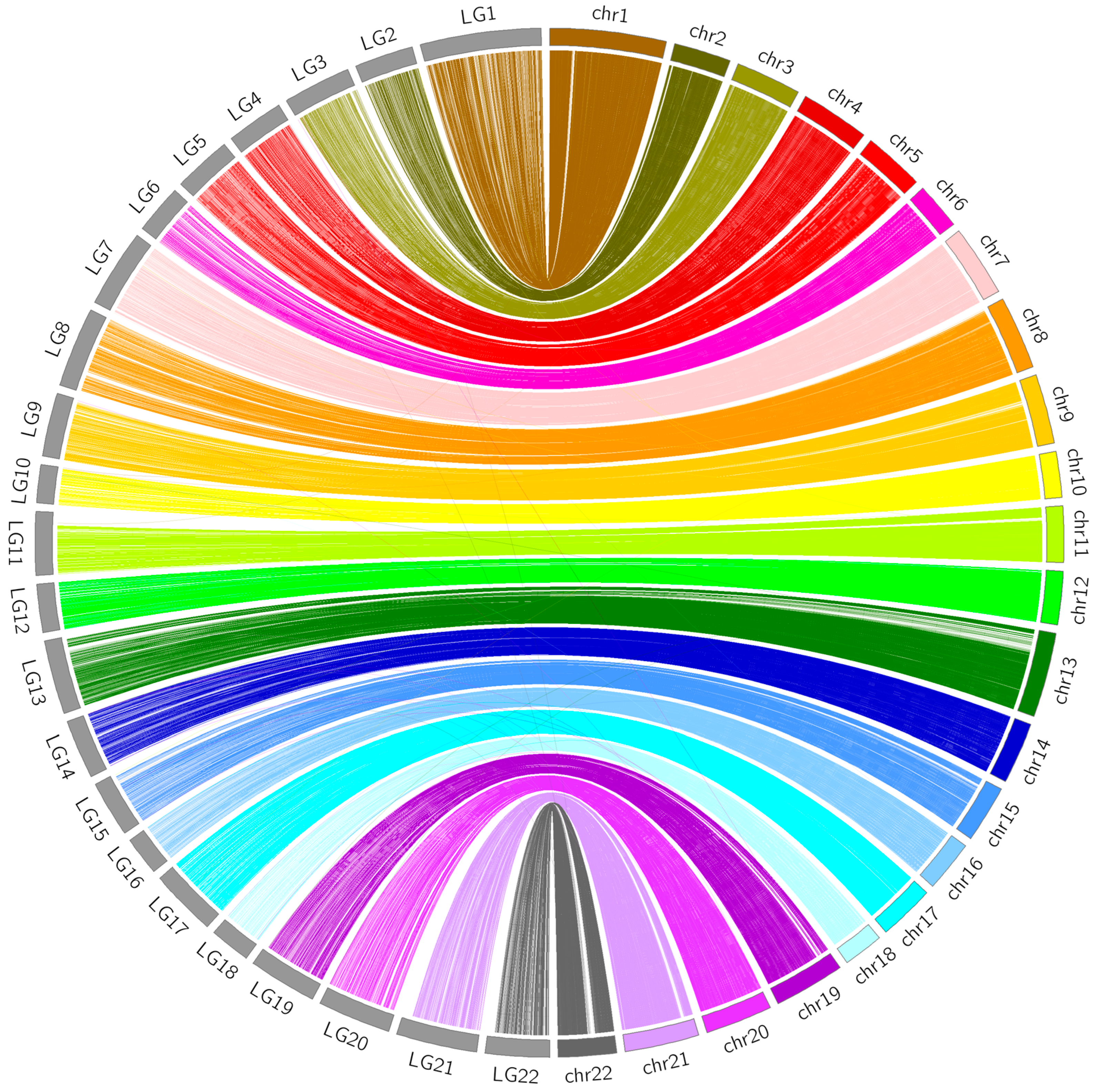
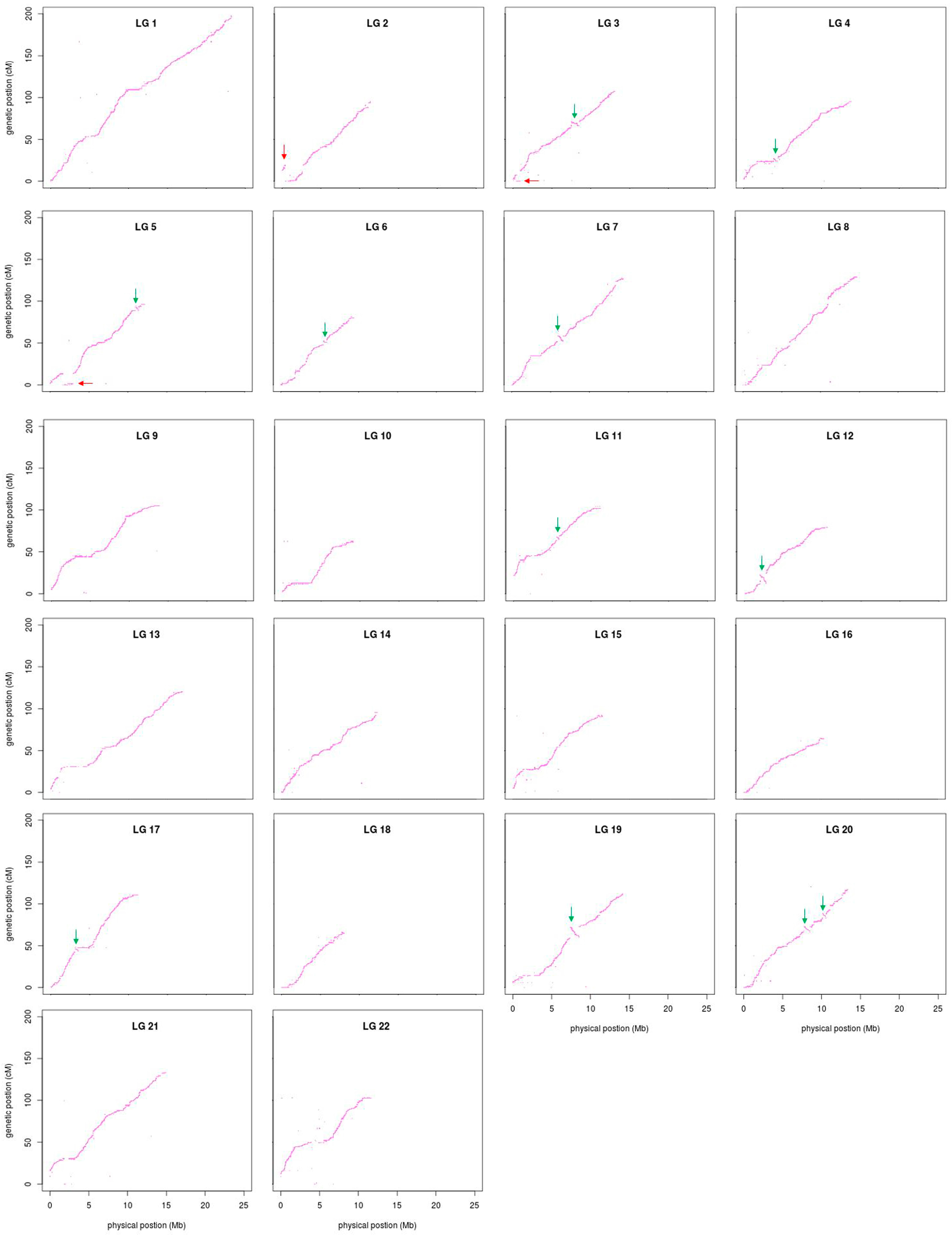
2.6. Comparative Analyses between the Linkage Map and FUGU5
3. Results
3.1. De Novo Assembly
3.2. Genetic Markers Genotyping
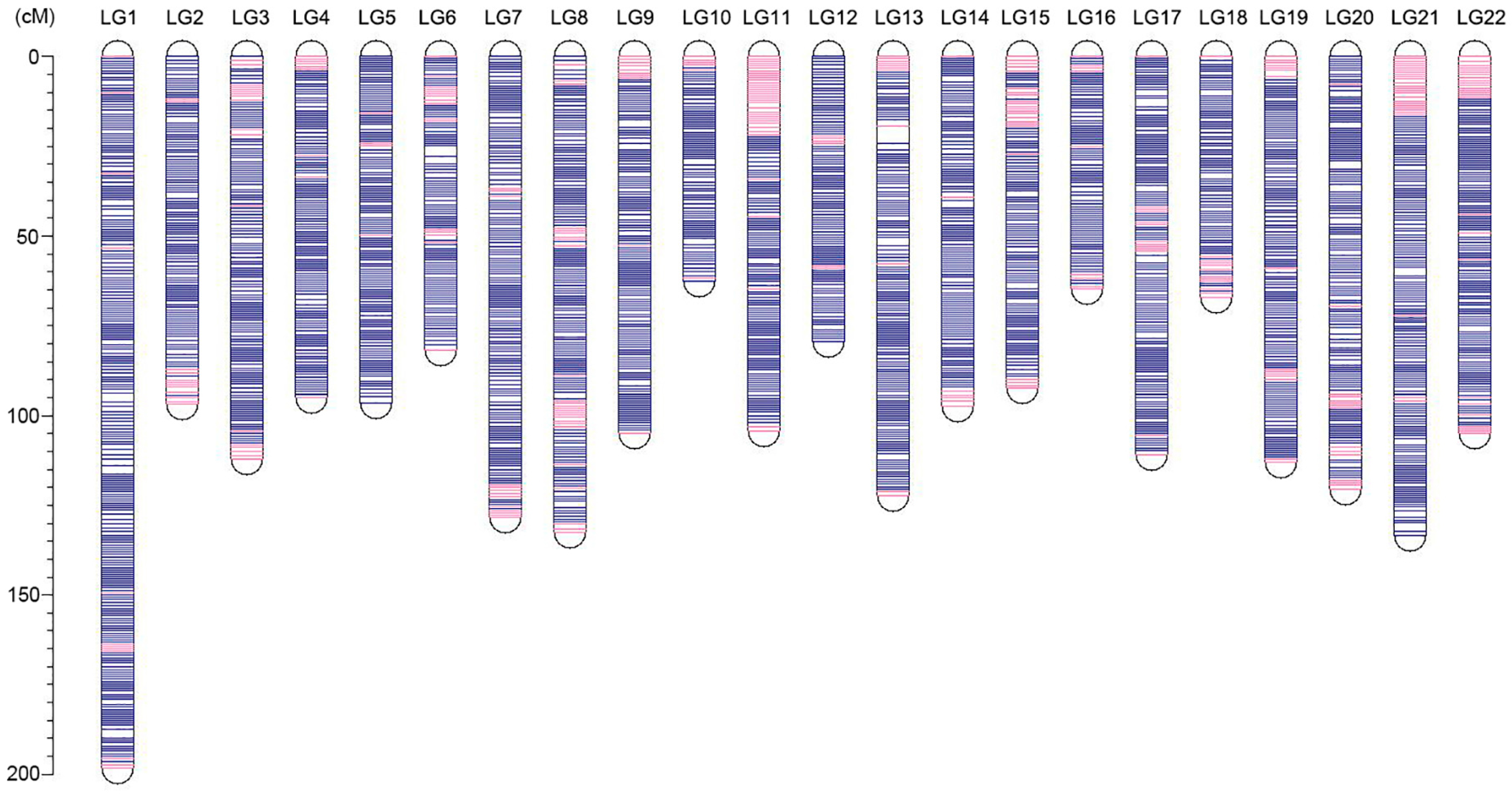
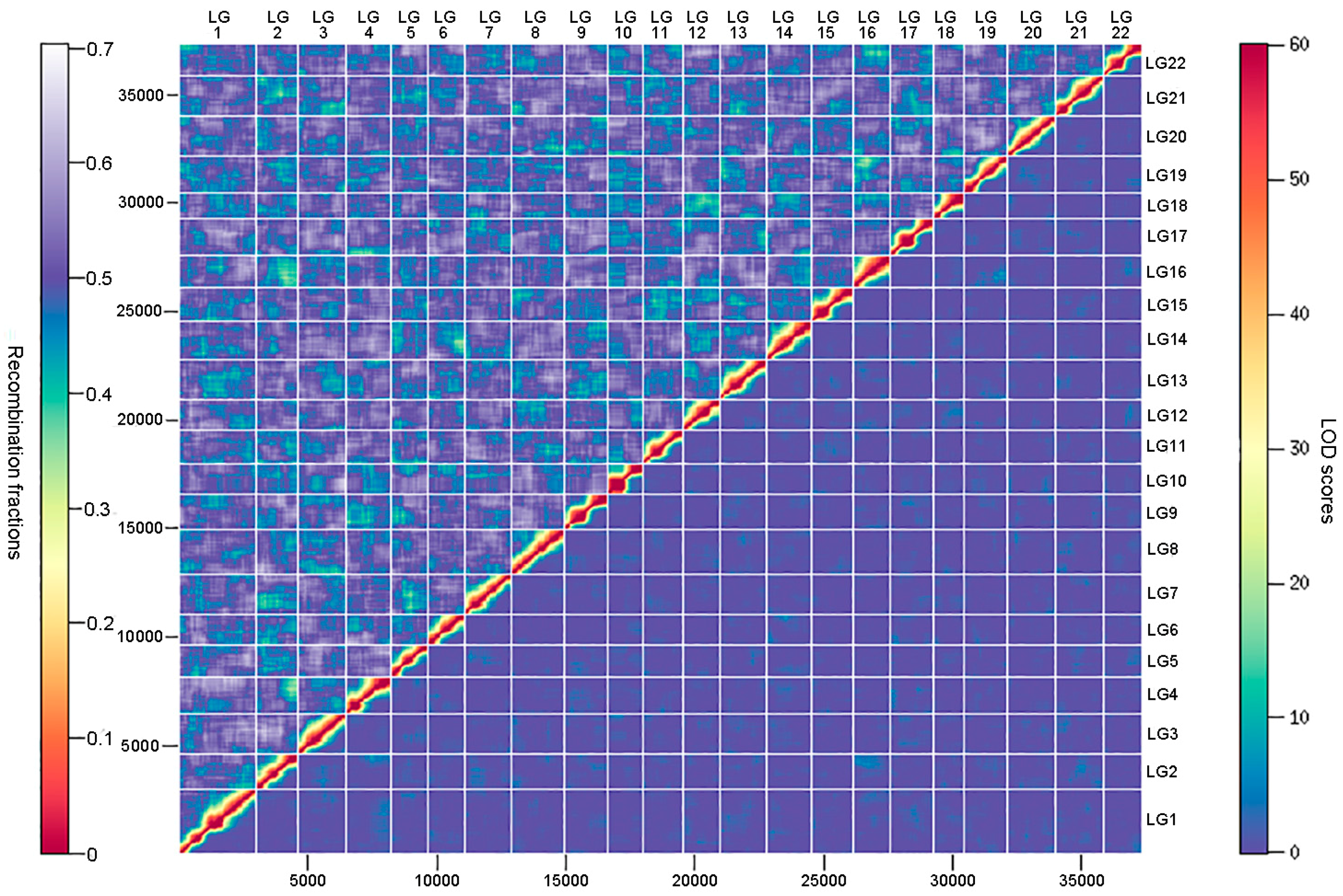
3.3. Ultrahigh-Density Genetic Linkage Map Construction for Torafugu
3.4. Comparative Analyses
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Burt, D.W. Comparative mapping in farm animals. Brief Funct. Genomic. Proteomic. 2002, 1, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Mun, J.H.; Kim, D.J.; Zhu, H.; Baek, J.M.; Mudge, J.; Roe, B.; Ellis, N.; Doyle, J.; Kiss, G.B.; et al. Estimating genome conservation between crop and model legume species. Proc. Natl. Acad. Sci. USA 2004, 101, 15289–15294. [Google Scholar] [CrossRef] [PubMed]
- Naruse, K.; Fukamachi, S.; Mitani, H.; Kondo, M.; Matsuoka, T.; Kondo, S.; Hanamura, N.; Morita, Y.; Hasegawa, K.; Nishigaki, R.; et al. A detailed linkage map of medaka, Oryzias latipes: Comparative genomics and genome evolution. Genetics 2000, 154, 1773–1784. [Google Scholar] [PubMed]
- Hollenbeck, C.M.; Portnoy, D.S.; Gold, J.R. A genetic linkage map of red drum (Sciaenops ocellatus) and comparison of chromosomal syntenies with four other fish species. Aquaculture 2015, 435, 265–274. [Google Scholar] [CrossRef]
- Kujur, A.; Upadhyaya, H.D.; Shree, T.; Bajaj, D.; Das, S.; Saxena, M.S.; Badoni, S.; Kumar, V.; Tripathi, S.; Gowda, C.L.; et al. Ultra-high density intra-specific genetic linkage maps accelerate identification of functionally relevant molecular tags governing important agronomic traits in chickpea. Sci. Rep. 2015, 5, 9468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Niu, Y.; Ye, X.; Dong, J.; Hu, W.; Zeng, Q.; Chen, Z.; Tian, Y.; Zhang, J.; Lu, M. Construction of a high-density linkage map and mapping of sex determination and growth-related loci in the mandarin fish (Siniperca chuatsi). BMC Genomics 2017, 18, 446. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, L.T.; Li, M.; Khan, M.A.; Li, X.G.; Chen, H.; Yin, H.; Zhang, S.L. High-density genetic linkage map construction and identification of fruit-related QTL in pear using SNP and SSR markers. J. Exp. Bot. 2014, 65, 5771–5781. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- International Barley Genome Sequencing Consortium; Mayer, K.F.; Waugh, R.; Brown, J.W.; Schulman, A.; Langridge, P.; Platzer, M.; Fincher, G.B.; Muehlbauer, G.J.; Sato, K.; et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Bao, B.; Xie, Z.; Chen, X.; Li, B.; Jia, X.; Yao, Q.; Orti, G.; Li, W.; Li, X.; et al. The genome and transcriptome of Japanese flounder provide insights into flatfish asymmetry. Nat. Genet. 2017, 49, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, Y.; Zhang, Y.; Ning, Z.; Li, Y.; Zhao, Q.; Lu, H.; Huang, R.; Xia, X.; Feng, Q.; et al. The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nat. Genet. 2015, 47, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Feng, Q.; Yu, H.; Huang, X.; Zhao, Q.; Xing, Y.; Yu, S.; Han, B.; Zhang, Q. Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 10578–10583. [Google Scholar] [CrossRef] [PubMed]
- Au, C.H.; Cheung, M.K.; Wong, M.C.; Chu, A.K.; Law, P.T.; Kwan, H.S. Rapid genotyping by low-coverage resequencing to construct genetic linkage maps of fungi: A case study in Lentinula edodes. BMC Res. Notes 2013, 6, 307. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.E.; Pearl, S.A.; Burke, J.M. Genetic mapping of millions of SNPs in safflower (Carthamus tinctorius L.) via whole-genome resequencing. G3-Genes Genom. Genet. 2016, 6, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Palloix, A.; Caranta, C.; Pochard, E. Construction of an intraspecific integrated linkage map of pepper using molecular markers and doubled haploid progenies. Genome 1995, 38, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Semagn, K.; Bjornstad, A.; Skinnes, H.; Maroy, A.G.; Tarkegne, Y.; William, M. Distribution of DArT, AFLP, and SSR markers in a genetic linkage map of a doubled-haploid hexaploid wheat population. Genome 2006, 49, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, W.; Zhang, T. Molecular linkage map of allotetraploid cotton (Gossypium hirsutum L. x Gossypium barbadense L.) with a haploid population. Theor. Appl. Genet. 2002, 105, 1166–1174. [Google Scholar] [PubMed]
- Xu, Y.; Huang, L.; Ji, D.; Chen, C.; Zheng, H.; Xie, C. Construction of a dense genetic linkage map and mapping quantitative trait loci for economic traits of a doubled haploid population of Pyropia haitanensis (Bangiales, Rhodophyta). BMC Plant Biol. 2015, 15, 228. [Google Scholar] [CrossRef] [PubMed]
- Delourme, R.; Falentin, C.; Fomeju, B.F.; Boillot, M.; Lassalle, G.; Andre, I.; Duarte, J.; Gauthier, V.; Lucante, N.; Marty, A.; et al. High-density SNP-based genetic map development and linkage disequilibrium assessment in Brassica napus L. BMC Genomics 2013, 14, 120. [Google Scholar] [CrossRef] [PubMed]
- Young, W.P.; Wheeler, P.A.; Coryell, V.H.; Keim, P.; Thorgaard, G.H. A detailed linkage map of rainbow trout produced using doubled haploids. Genetics 1998, 148, 839–850. [Google Scholar] [PubMed]
- Brieuc, M.S.; Waters, C.D.; Seeb, J.E.; Naish, K.A. A dense linkage map for Chinook salmon (Oncorhynchus tshawytscha) reveals variable chromosomal divergence after an ancestral whole genome duplication event. G3-Genes Genom. Genet. 2014, 4, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.D.; Chu, F.; Woods, I.G.; Ngo-Hazelett, P.; Cardozo, T.; Huang, H.; Kimm, F.; Liao, L.Y.; Yan, Y.L.; Zhou, Y.Y.; et al. Genetic linkage mapping of zebrafish genes and ESTs. Genome Res. 2000, 10, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.W.; Liang, L.Q. A genetic linkage map of common carp (Cyprinus carpio L.) and mapping of a locus associated with cold tolerance. Aquaculture 2004, 238, 165–172. [Google Scholar] [CrossRef]
- Komen, H.; Thorgaard, G.H. Androgenesis, gynogenesis and the production of clones in fishes: A review. Aquaculture 2007, 269, 150–173. [Google Scholar] [CrossRef]
- Dunwell, J.M. Haploids in flowering plants: Origins and exploitation. Plant Biotechnol. J. 2010, 8, 377–424. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.; Elgar, G.; Sandford, R.; Macrae, A.; Venkatesh, B.; Aparicio, S. Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature 1993, 366, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Grutzner, F.; Lutjens, G.; Rovira, C.; Barnes, D.W.; Ropers, H.H.; Haaf, T. Classical and molecular cytogenetics of the pufferfish Tetraodon nigroviridis. Chromosome Res. 1999, 7, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.B.; Kumar, S. Vertebrate genomes compared. Science 2002, 297, 1283–1285. [Google Scholar] [CrossRef] [PubMed]
- Kai, W.; Kikuchi, K.; Tohari, S.; Chew, A.K.; Tay, A.; Fujiwara, A.; Hosoya, S.; Suetake, H.; Naruse, K.; Brenner, S.; et al. Integration of the genetic map and genome assembly of fugu facilitates insights into distinct features of genome evolution in teleosts and mammals. Genome Biol. Evol. 2011, 3, 424–442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hirose, Y.; Yoshino, R.; Kinoshita, S.; Saito, K.; Tan, E.K.; Suzuki, Y.; Shimizu, A.; Okano, H.; Kudoh, J.; et al. Assessment of homozygosity levels in the mito-gynogenetic torafugu (Takifugu rubripes) by genome-wide SNP analyses. Aquaculture 2013, 380, 114–119. [Google Scholar] [CrossRef]
- Luo, R.B.; Liu, B.H.; Xie, Y.L.; Li, Z.Y.; Huang, W.H.; Yuan, J.Y.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Gadau, J. Phase-unknown linkage mapping in ants. Cold Spring Harb. Protoc. 2009, pdb.prot5251. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Bhat, P.R.; Close, T.J.; Lonardi, S. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet. 2008, 4, e1000212. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Butler, D. R package ASMap: Efficient genetic linkage map construction and diagnosis. J. Stat. Softw. 2017, 79, 1–29. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.J.; Zong, C.H.; Fan, W.; Yang, M.Y.; Li, J.S.; Chapman, A.R.; Zhu, P.; Hu, X.S.; Xu, L.Y.; Yan, L.Y.; et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 2012, 338, 1627–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Ackerman, M.S.; Long, H.A.; Bright, L.; Spitze, K.; Ramsdell, J.S.; Thomas, W.K.; Lynch, M. A male-specific genetic map of the microcrustacean Daphnia pulex based on single-sperm whole-genome sequencing. Genetics 2015, 201, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, C.S.; Newnham, L.J.; Capalbo, A.; Natesan, S.A.; Joshi, H.A.; Cimadomo, D.; Griffin, D.K.; Sage, K.; Summers, M.C.; Thornhill, A.R.; et al. Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat. Genet. 2015, 47, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, C.S.; Capalbo, A.; Newnham, L.; Cimadomo, D.; Natesan, S.A.; Hoffmann, E.R.; Ubaldi, F.M.; Rienzi, L.; Handyside, A.H. Generation of meiomaps of genome-wide recombination and chromosome segregation in human oocytes. Nat. Protoc. 2016, 11, 1229–1243. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.; Pollara, V.J.; Cowles, C.R.; Van Etten, W.J.; Baldwin, J.; Linton, L.; Lander, E.S. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 2000, 407, 513–516. [Google Scholar] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkage Group | Genetic Distance (cM) | SSGs Numbers | SNPs Numbers | Unique Positions | Marker Interval |
|---|---|---|---|---|---|
| LG1 | 198.25 | 2997 | 64,119 | 252 | 0.79 |
| LG2 | 96.95 | 1588 | 34,369 | 128 | 0.76 |
| LG3 | 112.13 | 1876 | 39,280 | 147 | 0.76 |
| LG4 | 95.15 | 1722 | 35,535 | 132 | 0.72 |
| LG5 | 96.42 | 1454 | 31,043 | 136 | 0.71 |
| LG6 | 81.72 | 1431 | 31,616 | 110 | 0.74 |
| LG7 | 128.42 | 1819 | 37,225 | 166 | 0.77 |
| LG8 | 132.44 | 2055 | 44,302 | 174 | 0.76 |
| LG9 | 104.88 | 1670 | 34,975 | 143 | 0.73 |
| LG10 | 62.75 | 1396 | 30,320 | 82 | 0.77 |
| LG11 | 104.43 | 1543 | 33,186 | 143 | 0.73 |
| LG12 | 79.40 | 1408 | 30,636 | 110 | 0.72 |
| LG13 | 122.28 | 1797 | 38,739 | 157 | 0.78 |
| LG14 | 97.47 | 1770 | 37,750 | 128 | 0.76 |
| LG15 | 92.43 | 1605 | 35,558 | 119 | 0.78 |
| LG16 | 64.71 | 1417 | 30,875 | 89 | 0.73 |
| LG17 | 110.91 | 1727 | 38,620 | 150 | 0.74 |
| LG18 | 67.20 | 1157 | 26,774 | 89 | 0.76 |
| LG19 | 112.95 | 1714 | 35,312 | 154 | 0.73 |
| LG20 | 120.46 | 1830 | 38,989 | 152 | 0.79 |
| LG21 | 133.31 | 1893 | 40,975 | 180 | 0.74 |
| LG22 | 104.98 | 1474 | 32,079 | 149 | 0.70 |
| Total | 2319.65 | 37,343 | 802,277 | 3090 | 0.75 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Mizukoshi, M.; Zhang, H.; Tan, E.; Igarashi, Y.; Suzuki, Y.; Mitsuyama, S.; Kinoshita, S.; Saito, K.; Watabe, S.; et al. Ultrahigh-Density Linkage Map Construction Using Low-Coverage Whole-Genome Sequencing of a Doubled Haploid Population: Case Study of Torafugu (Takifugu rubripes). Genes 2018, 9, 120. https://doi.org/10.3390/genes9030120
Zhang X, Mizukoshi M, Zhang H, Tan E, Igarashi Y, Suzuki Y, Mitsuyama S, Kinoshita S, Saito K, Watabe S, et al. Ultrahigh-Density Linkage Map Construction Using Low-Coverage Whole-Genome Sequencing of a Doubled Haploid Population: Case Study of Torafugu (Takifugu rubripes). Genes. 2018; 9(3):120. https://doi.org/10.3390/genes9030120
Chicago/Turabian StyleZhang, Xiang, Misaki Mizukoshi, Hong Zhang, Engkong Tan, Yoji Igarashi, Yutaka Suzuki, Susumu Mitsuyama, Shigeharu Kinoshita, Kazuyoshi Saito, Shugo Watabe, and et al. 2018. "Ultrahigh-Density Linkage Map Construction Using Low-Coverage Whole-Genome Sequencing of a Doubled Haploid Population: Case Study of Torafugu (Takifugu rubripes)" Genes 9, no. 3: 120. https://doi.org/10.3390/genes9030120
APA StyleZhang, X., Mizukoshi, M., Zhang, H., Tan, E., Igarashi, Y., Suzuki, Y., Mitsuyama, S., Kinoshita, S., Saito, K., Watabe, S., & Asakawa, S. (2018). Ultrahigh-Density Linkage Map Construction Using Low-Coverage Whole-Genome Sequencing of a Doubled Haploid Population: Case Study of Torafugu (Takifugu rubripes). Genes, 9(3), 120. https://doi.org/10.3390/genes9030120