Resolving the Enigma of the Clonal Expansion of mtDNA Deletions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Computer Simulations of mtDNA Accumulation
2.2. Databases
3. Results
4. Discussion
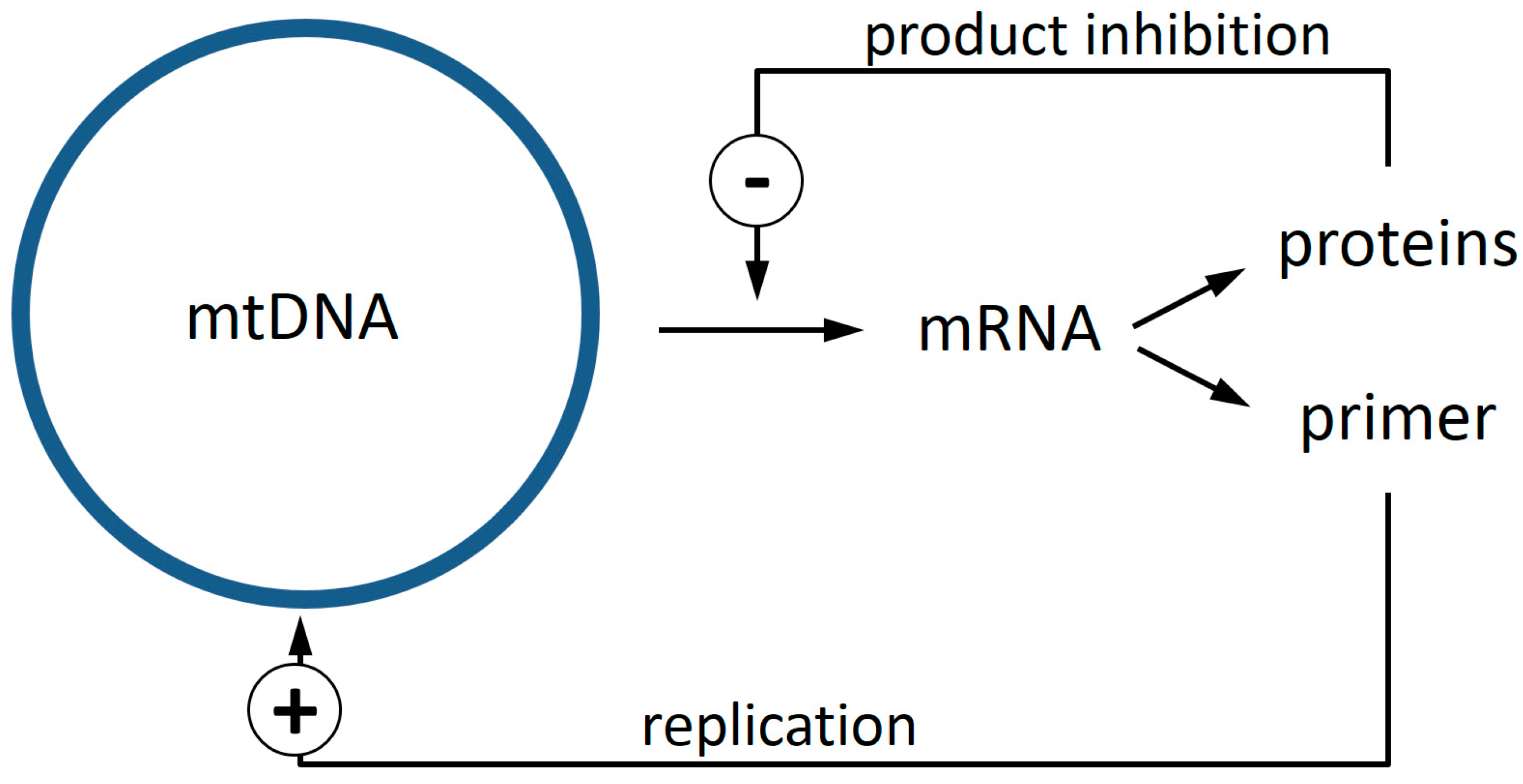
- It provides a biochemical mechanism that confers a selective advantage to defective mtDNA molecules.
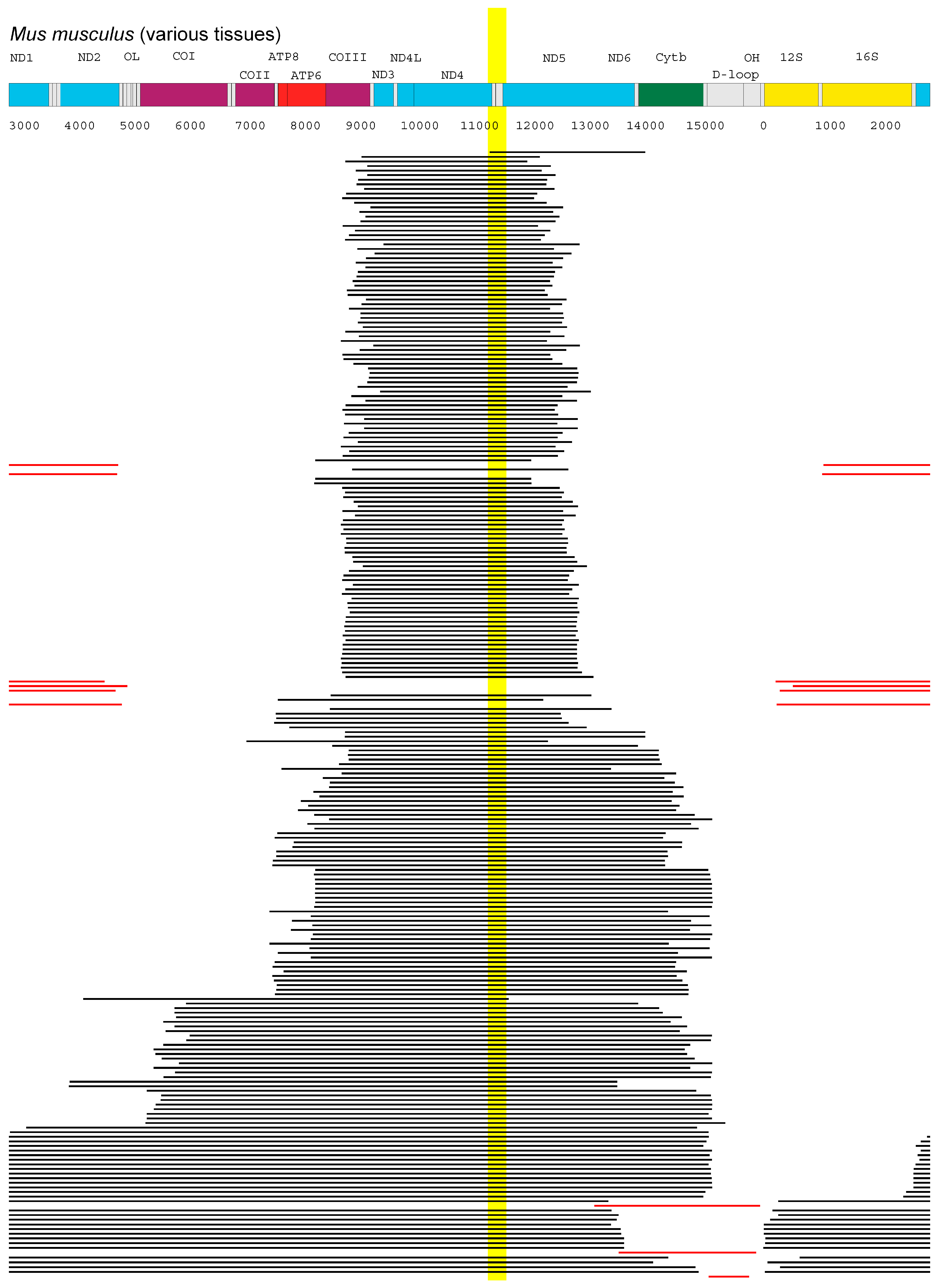
- Experimental data sets from mouse, rat, rhesus monkey, and human specimens all point to a region of mtDNA that is shared between most of the deletions. The genes of this region, ND4 and possibly ND5, are prime candidates for components of the proposed feedback mechanism.
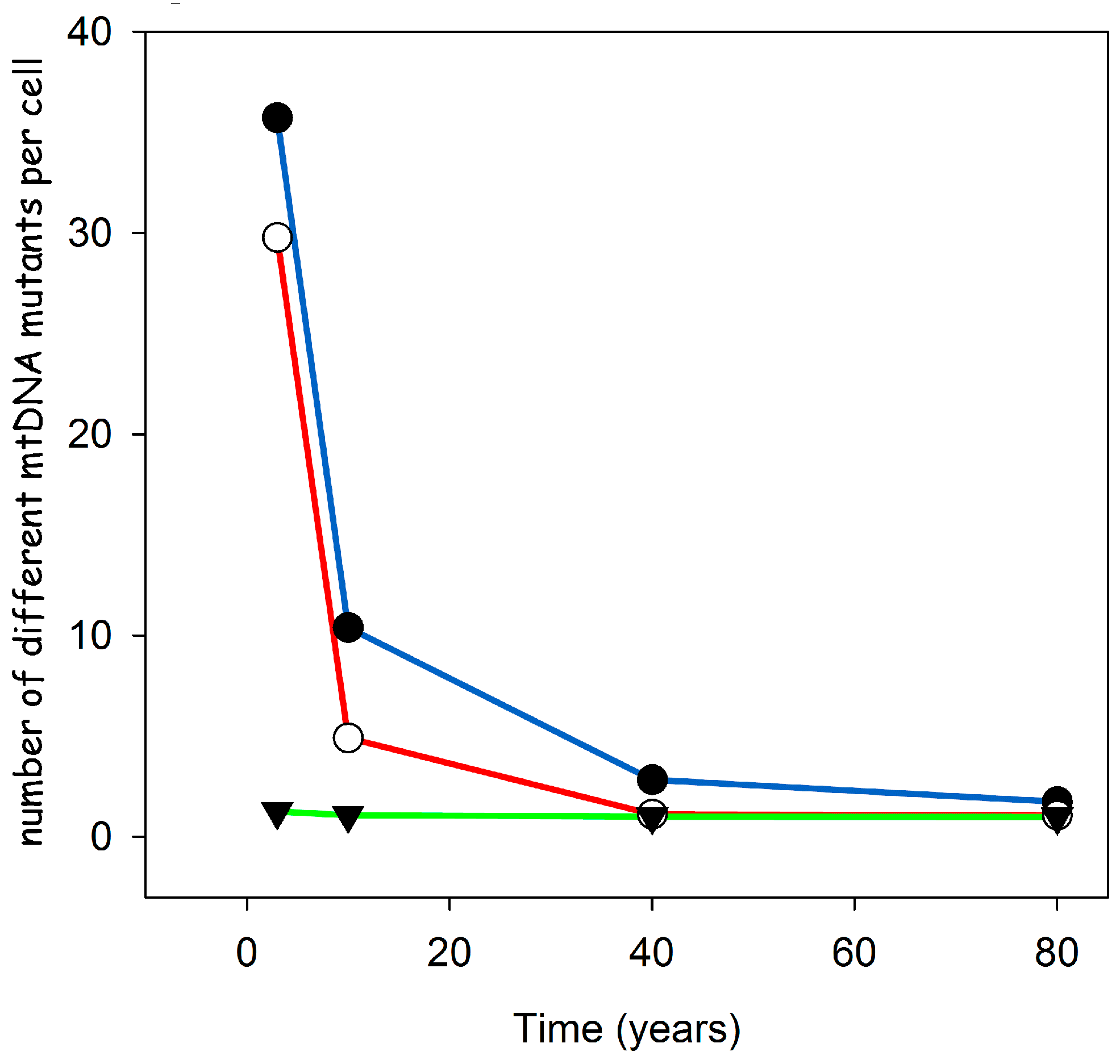
- Computer calculations show that the suggested mechanism leads to a very low level of heteroplasmy, as observed experimentally.
- Importantly, this low level of heteroplasmy is also predicted for short lived animals like mice and rats. This is in stark contrast to other ideas like random drift or size advantage.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kontis, V.; Bennett, J.E.; Mathers, C.D.; Li, G.; Foreman, K.; Ezzati, M. Future life expectancy in 35 industrialised countries: Projections with a bayesian model ensemble. Lancet 2017, 389, 1323–1335. [Google Scholar] [CrossRef]
- Cao, Z.; Wanagat, J.; McKiernan, S.H.; Aiken, J.M. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: Analysis by laser-capture microdissection. Nucleic Acids Res. 2001, 29, 4502–4508. [Google Scholar] [CrossRef] [PubMed]
- Gokey, N.G.; Cao, Z.; Pak, J.W.; Lee, D.; McKiernan, S.H.; McKenzie, D.; Weindruch, R.; Aiken, J.M. Molecular analyses of mtDNA deletion mutations in microdissected skeletal muscle fibers from aged rhesus monkeys. Aging Cell 2004, 3, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Brierley, E.J.; Johnson, M.A.; Lightowlers, R.N.; James, O.F.; Turnbull, D.M. Role of mitochondrial DNA mutations in human aging: Implications for the central nervous system and muscle. Ann. Neurol. 1998, 43, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Bodyak, N.; Thilly, W.G.; van Orsouw, N.J.; Zhang, X.; Coller, H.A.; Perls, T.T.; Upton, M.; Vijg, J.; Wei, J.Y. Cell by cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999, 27, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Pak, J.W.; McKenzie, D.; Bua, E.; Bassiouni, M.; Aiken, J.M. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: Evidence for a causal role in muscle fiber loss. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Barron, M.J.; Borthwick, G.M.; Gospel, A.; Chinnery, P.F.; Samuels, D.C.; Taylor, G.A.; Plusa, S.M.; Needham, S.J.; Greaves, L.C.; et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest. 2003, 112, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Barron, M.J.; Campbell-Shiel, G.; Kirkwood, T.B.; Turnbull, D.M. Differences in the accumulation of mitochondrial defects with age in mice and humans. Mech. Ageing Dev. 2011, 132, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Kirkwood, T.B. Mitochondrial mutations and aging: Random drift is insufficient to explain the accumulation of mitochondrial deletion mutants in short-lived animals. Aging Cell 2013, 12, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Kirkwood, T.B. Transcription could be the key to the selection advantage of mitochondrial deletion mutants in aging. Proc. Natl. Acad. Sci USA 2014, 111, 2972–2977. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Speijer, D.; Kirkwood, T.B. The road to rack and ruin: Selecting deleterious mitochondrial DNA variants. Philos. Trans. R Soc Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science 1992, 256, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Lopez, M.E.; Weindruch, R.; Aiken, J.M. Association of age-related mitochondrial abnormalities with skeletal muscle fiber atrophy. Free Radic. Biol. Med. 1998, 25, 964–972. [Google Scholar] [CrossRef]
- Diaz, F.; Bayona-Bafaluy, M.P.; Rana, M.; Mora, M.; Hao, H.; Moraes, C.T. Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res. 2002, 30, 4626–4633. [Google Scholar] [CrossRef] [PubMed]
- de Grey, A.D.N.J. A proposed refinement of the mitochondrial free radical theory of aging. Bioessays 1997, 19, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Dawson, M.; Kirkwood, T.B. Mitochondrial mutations and ageing: Can mitochondrial deletion mutants accumulate via a size based replication advantage? J. Theor. Biol. 2014, 340, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Kirkwood, T.B. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proc. Natl. Acad. Sci. USA 2011, 108, 10237–10242. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.F.; Narendra, D.P.; Tanaka, A.; Manfredi, G.; Youle, R.J. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11835–11840. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Lemasters, J.J. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid. Redox Signal. 2011, 14, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Silva, P.; Enriquez, J.A.; Montoya, J. Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 2003, 88, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Wolfram-Research, Inc. Mathematica, version 10; Wolfram Research, Inc.: Champaign, IL, USA, 2015. [Google Scholar]
- Damas, J.; Carneiro, J.; Amorim, A.; Pereira, F. Mitobreak: The mitochondrial DNA breakpoints database. Nucleic Acids Res. 2014, 42, D1261–D1268. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Krishnan, K.J.; Elson, J.L.; Morris, C.M.; Bender, A.; Lightowlers, R.N.; Turnbull, D.M. Nature of mitochondrial DNA deletions in substantia nigra neurons. Am. J. Hum. Genet. 2008, 82, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Wanagat, J.; Cao, Z.; Pathare, P.; Aiken, J.M. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 2001, 15, 322–332. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, D.; Bua, E.; McKiernan, S.; Cao, Z.; Aiken, J.M.; Jonathan, W. Mitochondrial DNA deletion mutations: A causal role in sarcopenia. Eur. J. Biochem. 2002, 269, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Arnheim, N.; Cortopassi, G. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutat. Res. 1992, 275, 157–167. [Google Scholar] [CrossRef]
- Bandy, B.; Davison, A.J. Mitochondrial mutations may increase oxidative stress: Implications for carcinogenesis and aging? Free Radic. Biol. Med. 1990, 8, 523–539. [Google Scholar] [CrossRef]
- Elson, J.L.; Samuels, D.C.; Turnbull, D.M.; Chinnery, P.F. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am. J. Hum. Genet. 2001, 68, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Samuels, D.C. Relaxed replication of mtDNA: A model with implications for the expression of disease. Am. J. Hum. Genet. 1999, 64, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.; Krishnan, K.J.; Deschauer, M.; Taylor, R.W.; Turnbull, D.M. Dissecting the mechanisms underlying the accumulation of mitochondrial DNA deletions in human skeletal muscle. Hum. Mol. Genet. 2014, 23, 4612–4620. [Google Scholar] [CrossRef] [PubMed]
- Gitschlag, B.L.; Kirby, C.S.; Samuels, D.C.; Gangula, R.D.; Mallal, S.A.; Patel, M.R. Homeostatic responses regulate selfish mitochondrial genome dynamics in C. elegans. Cell Metab. 2016, 24, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Moraes, C.T. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 2009, 18, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Gross, N.J.; Getz, G.S.; Rabinowitz, M. Apparent turnover of mitochondrial deoxyribonucleic acid and mitochondrial phospholipids in the tissues of the rat. J. Biol. Chem. 1969, 244, 1552–1562. [Google Scholar] [PubMed]
- Huemer, R.P.; Lee, K.D.; Reeves, A.E.; Bickert, C. Mitochondrial studies in senescent mice—II. Specific activity, bouyant density, and turnover of mitochondrial DNA. Exp. Gerontol. 1971, 6, 327–334. [Google Scholar] [CrossRef]
- Korr, H.; Kurz, C.; Seidler, T.O.; Sommer, D.; Schmitz, C. Mitochondrial DNA synthesis studied autoradiographically in various cell types in vivo. Braz. J. Med. Biol. Res. 1998, 31, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.L.; Eng, S.; Hoh, R.; Hellerstein, M.K. Measurement of mitochondrial DNA synthesis in vivo using a stable isotope-mass spectrometric technique. J. Appl. Physiol. 2003, 94, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, A.; Kraytsberg, Y.; Guo, X.; Khrapko, K. On the timing and the extent of clonal expansion of mtDNA deletions: Evidence from single-molecule PCR. Exp. Neurol. 2009, 218, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K. The timing of mitochondrial DNA mutations in aging. Nat. Genet. 2011, 43, 726–727. [Google Scholar] [CrossRef] [PubMed]
- Popadin, K.; Safdar, A.; Kraytsberg, Y.; Khrapko, K. When man got his mtDNA deletions? Aging Cell 2014, 13, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, S.K.; Hance, N.; El Meziane, A.; Juhola, M.K.; Juhola, K.M.; Karhu, R.; Spelbrink, J.N.; Holt, I.J.; Jacobs, H.T. Genotypic stability, segregation and selection in heteroplasmic human cell lines containing np 3243 mutant mtDNA. Genetics 2000, 154, 363–380. [Google Scholar] [PubMed]
- Yoneda, M.; Chomyn, A.; Martinuzzi, A.; Hurko, O.; Attardi, G. Marked replicative advantage of human mtDNA carrying a point mutation that causes the melas encephalomyopathy. Proc. Natl. Acad. Sci. USA 1992, 89, 11164–11168. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Wanagat, J.; Cheema, N.; Widjaja, K.; McKenzie, D.; Aiken, J.M. Latent mitochondrial DNA deletion mutations drive muscle fiber loss at old age. Aging Cell 2016, 15, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Mouli, P.K.; Twig, G.; Shirihai, O.S. Frequency and selectivity of mitochondrial fusion are key to its quality maintenance function. Biophys. J. 2009, 96, 3509–3518. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Popadin, K.Y.; Markuzon, N.; Orlov, Y.L.; Kraytsberg, Y.; Krishnan, K.J.; Zsurka, G.; Turnbull, D.M.; Kunz, W.S.; Khrapko, K. Repeats, longevity and the sources of mtDNA deletions: Evidence from ‘deletional spectra’. Trends Genet. 2010, 26, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, L.N.; Gruber, J.; Halliwell, B.; Gunawan, R. Role of direct repeat and stem-loop motifs in mtDNA deletions: Cause or coincidence? PLoS ONE 2012, 7, e35271. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.N.; Seluanov, A.; Gorbunova, V. Mitochondrial inverted repeats strongly correlate with lifespan: mtDNA inversions and aging. PLoS ONE 2013, 8, e73318. [Google Scholar] [CrossRef] [PubMed]
- Tam, Z.Y.; Gruber, J.; Halliwell, B.; Gunawan, R. Context-dependent role of mitochondrial fusion-fission in clonal expansion of mtDNA mutations. PLoS Comput. Biol. 2015, 11, e1004183. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zheng, K.; Clark, J.; Swerdlow, R.H.; Pulst, S.M.; Sutton, J.P.; Shinobu, L.A.; Simon, D.K. Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum. Mol. Genet. 2013, 6, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Kandul, N.P.; Zhang, T.; Hay, B.A.; Guo, M. Selective removal of deletion-bearing mitochondrial DNA in heteroplasmic Drosophila. Nature Commun. 2016, 7, 13100. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowald, A.; Kirkwood, T.B.L. Resolving the Enigma of the Clonal Expansion of mtDNA Deletions. Genes 2018, 9, 126. https://doi.org/10.3390/genes9030126
Kowald A, Kirkwood TBL. Resolving the Enigma of the Clonal Expansion of mtDNA Deletions. Genes. 2018; 9(3):126. https://doi.org/10.3390/genes9030126
Chicago/Turabian StyleKowald, Axel, and Thomas B.L. Kirkwood. 2018. "Resolving the Enigma of the Clonal Expansion of mtDNA Deletions" Genes 9, no. 3: 126. https://doi.org/10.3390/genes9030126
APA StyleKowald, A., & Kirkwood, T. B. L. (2018). Resolving the Enigma of the Clonal Expansion of mtDNA Deletions. Genes, 9(3), 126. https://doi.org/10.3390/genes9030126