Testing for Polytomies in Phylogenetic Species Trees Using Quartet Frequencies


Abstract
:1. Introduction
2. Materials and Methods
2.1. Background
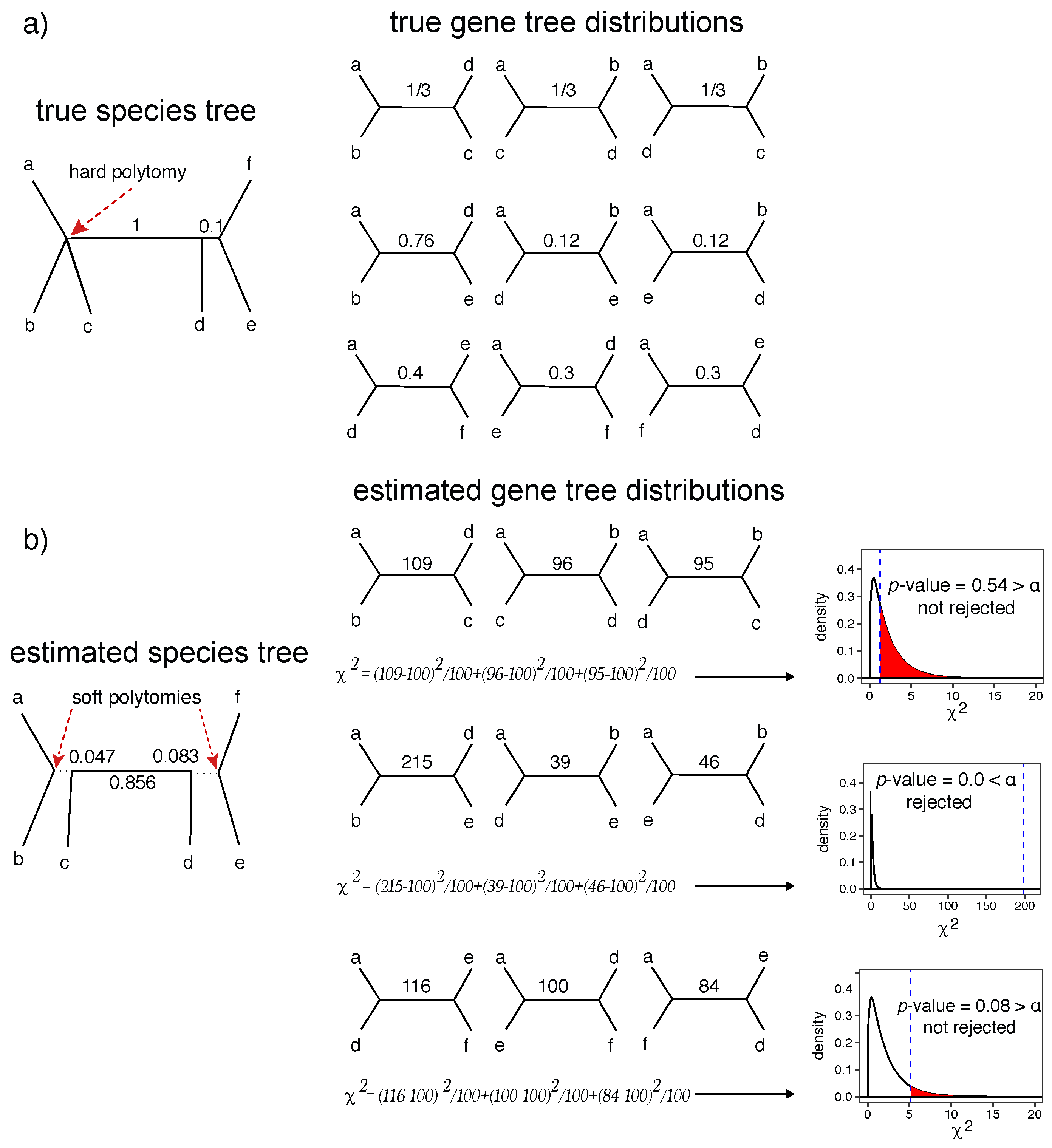
2.2. A Statistical Test of Polytomy
- Null hypothesis:
- The length of the internal branch is zero; thus, the species tree has a polytomy.
- A1.
- All positive length branches in the given species tree are correct.
- A2.
- Gene trees are a random error-free sample from the distribution defined by the MSC model.
- A3.
- We have gene trees with at least a quartet relevant to .
2.3. Evaluations
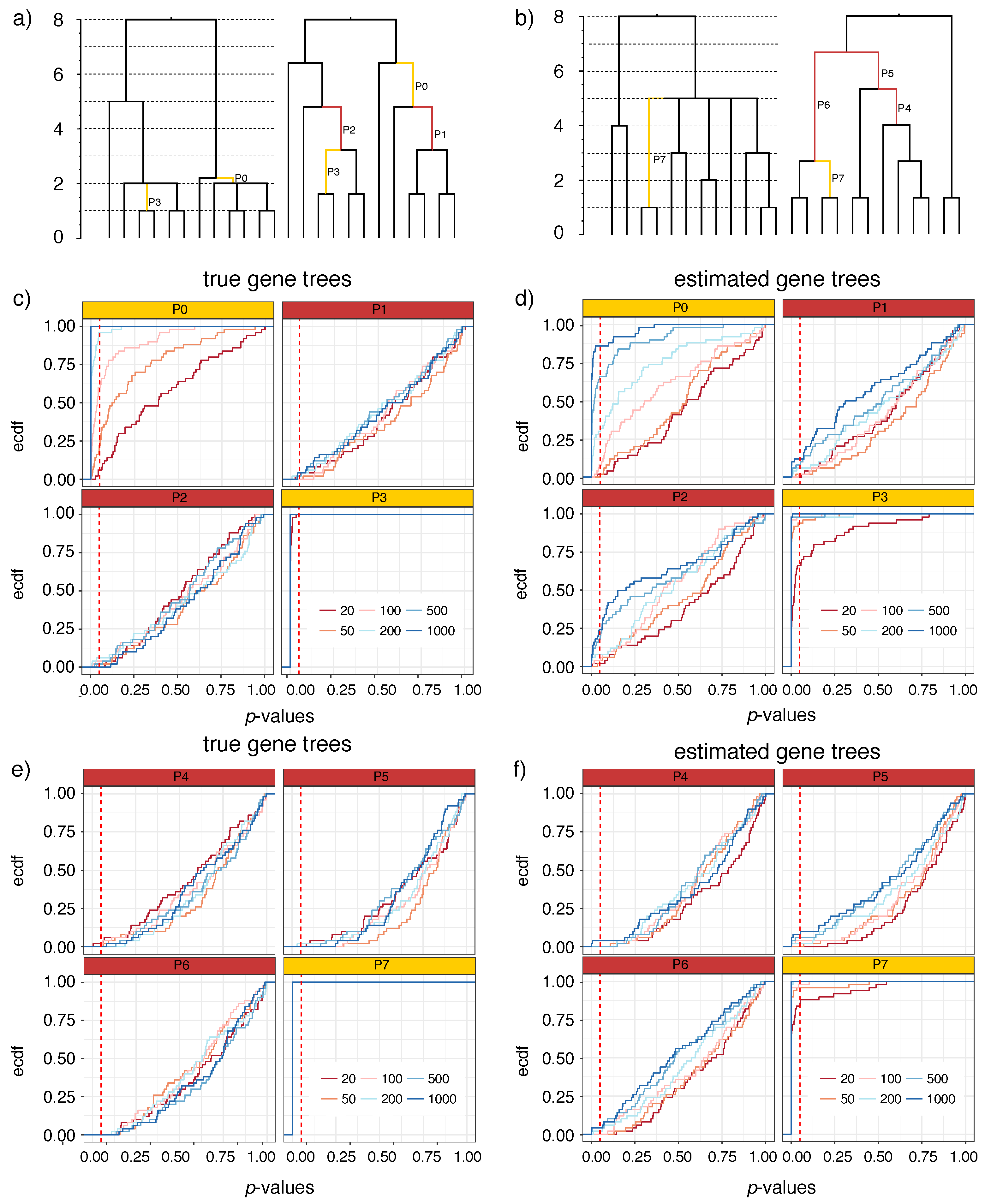
2.3.1. Simulated Datasets with Polytomies (S12A and S12B)
2.3.2. Simulated Datasets without Polytomies (S201)
3. Results
3.1. Simulated Datasets
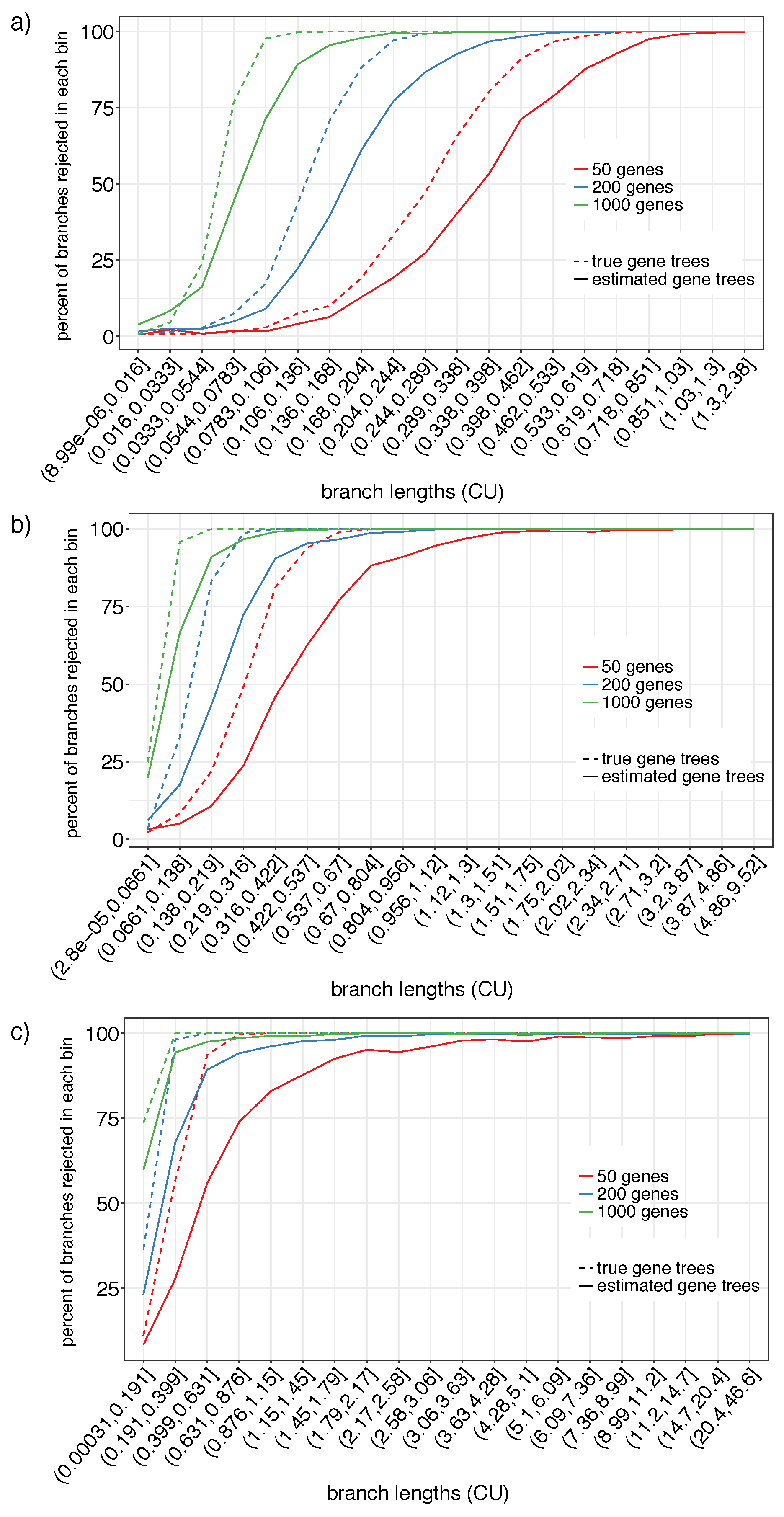
3.1.1. S12A and S12B
3.1.2. S201
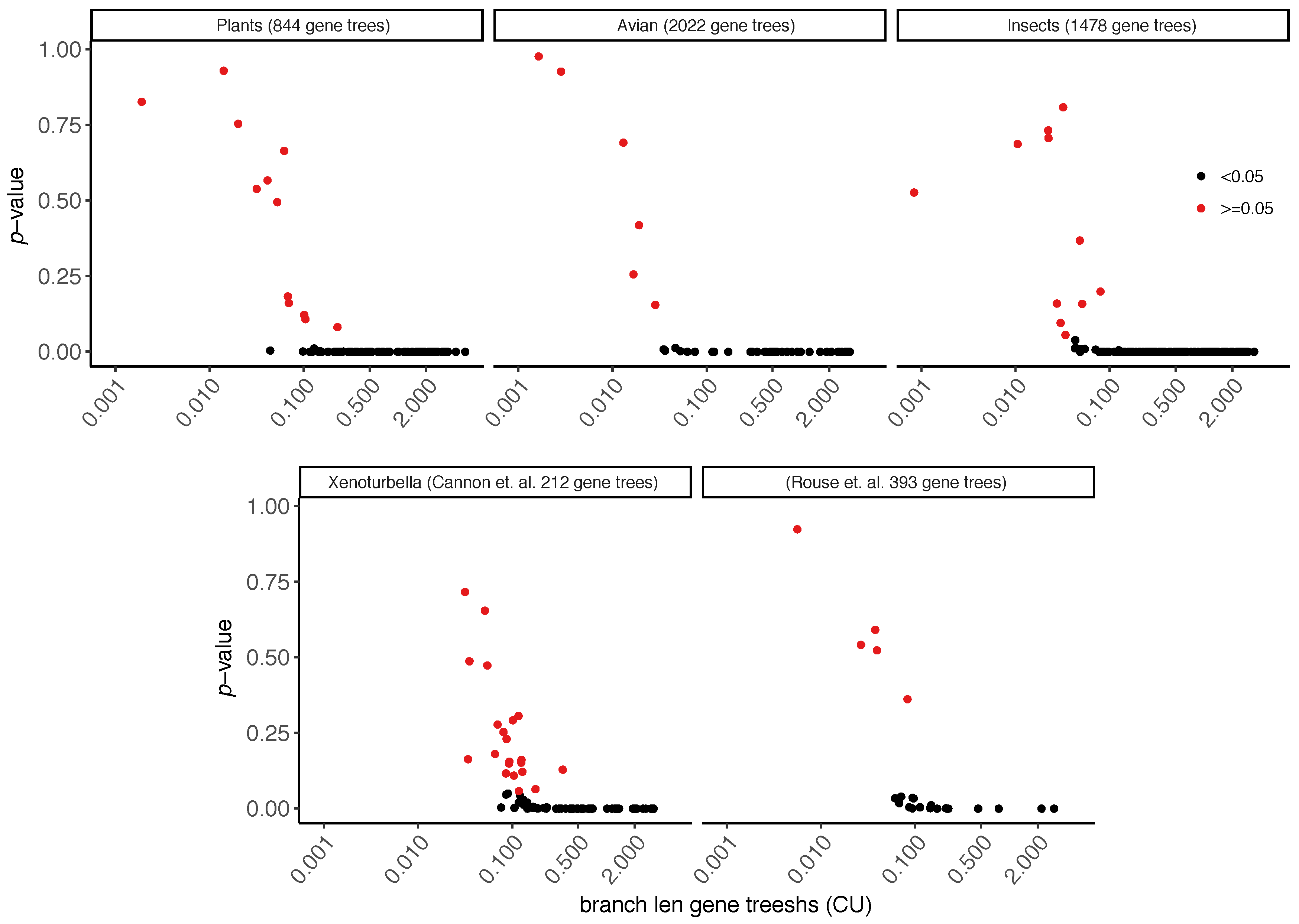
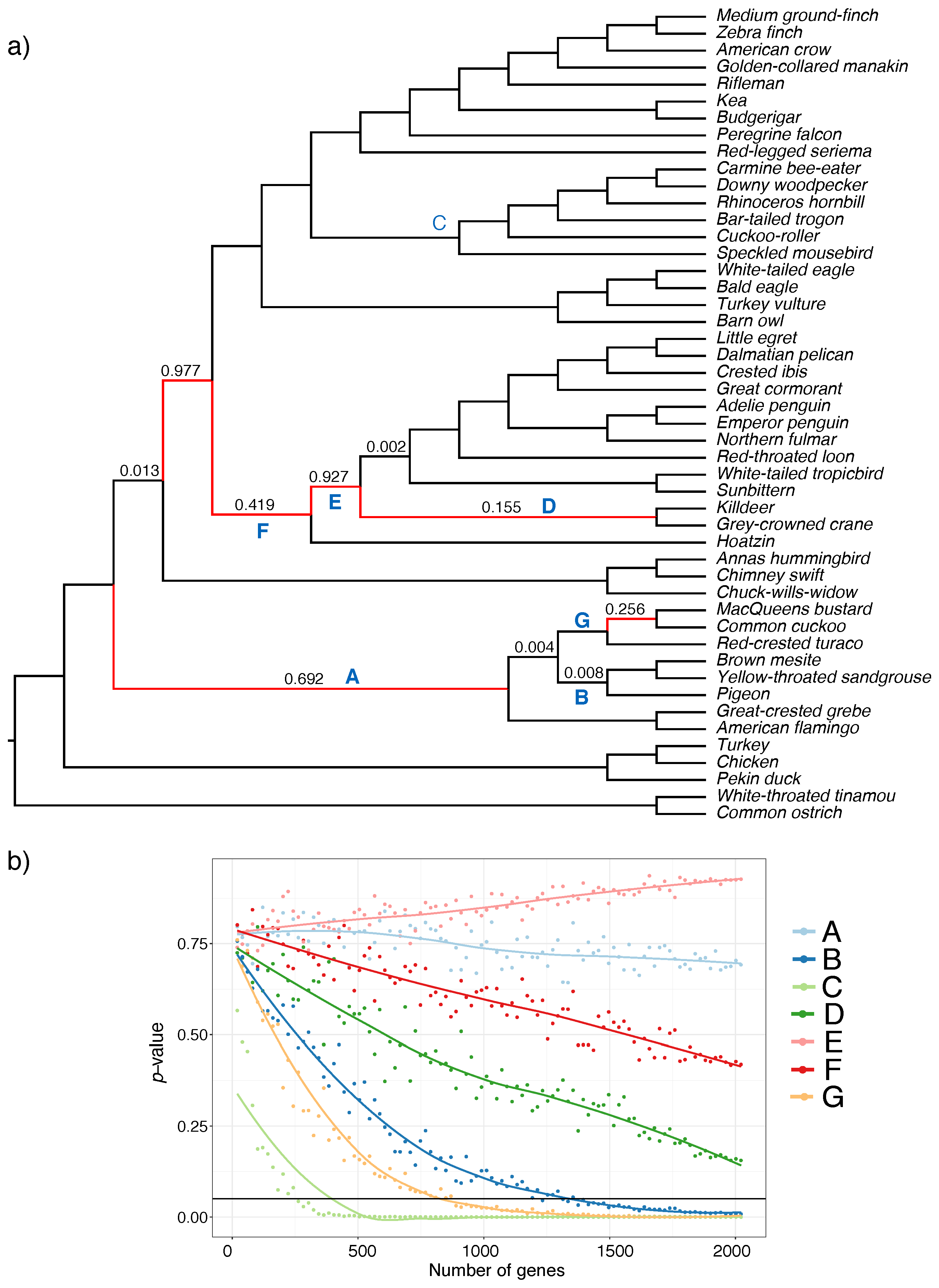
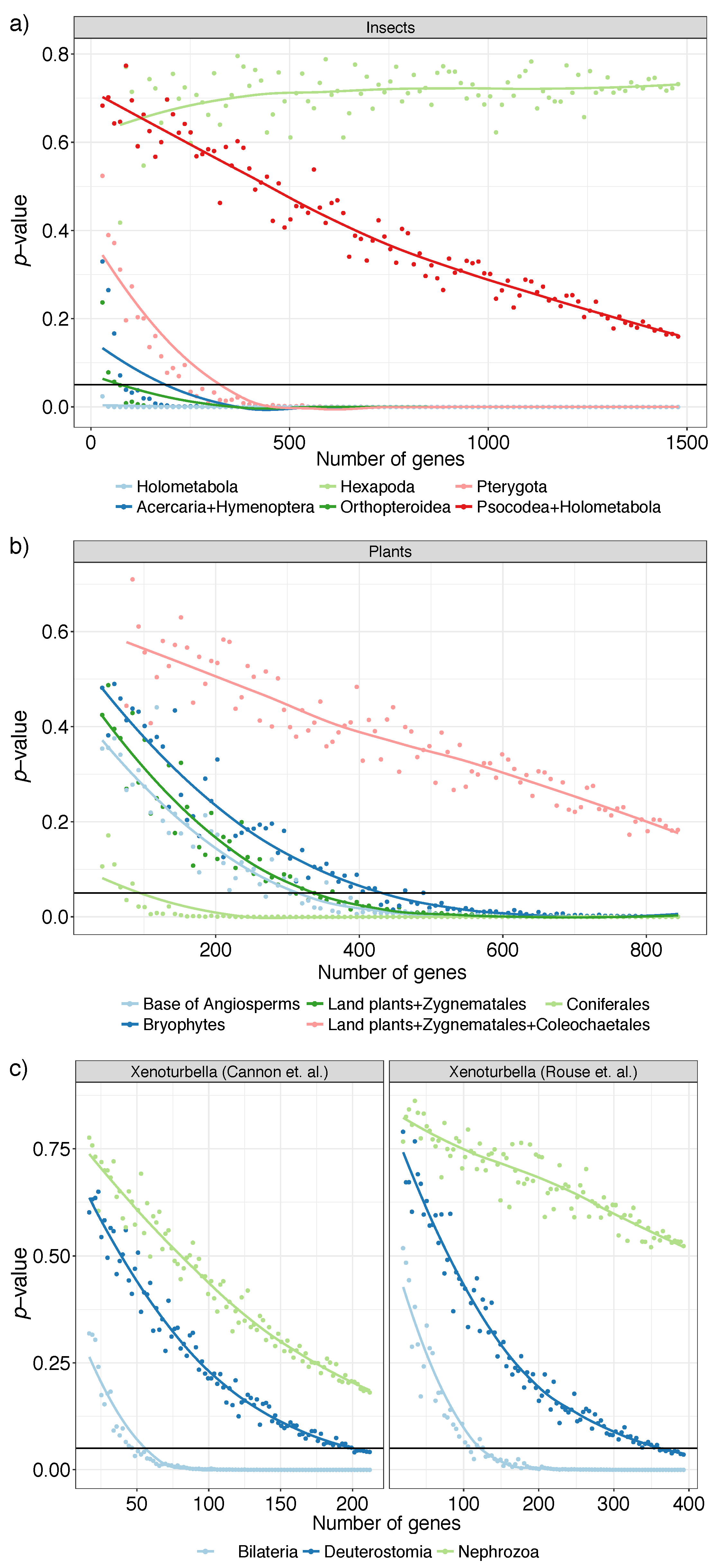
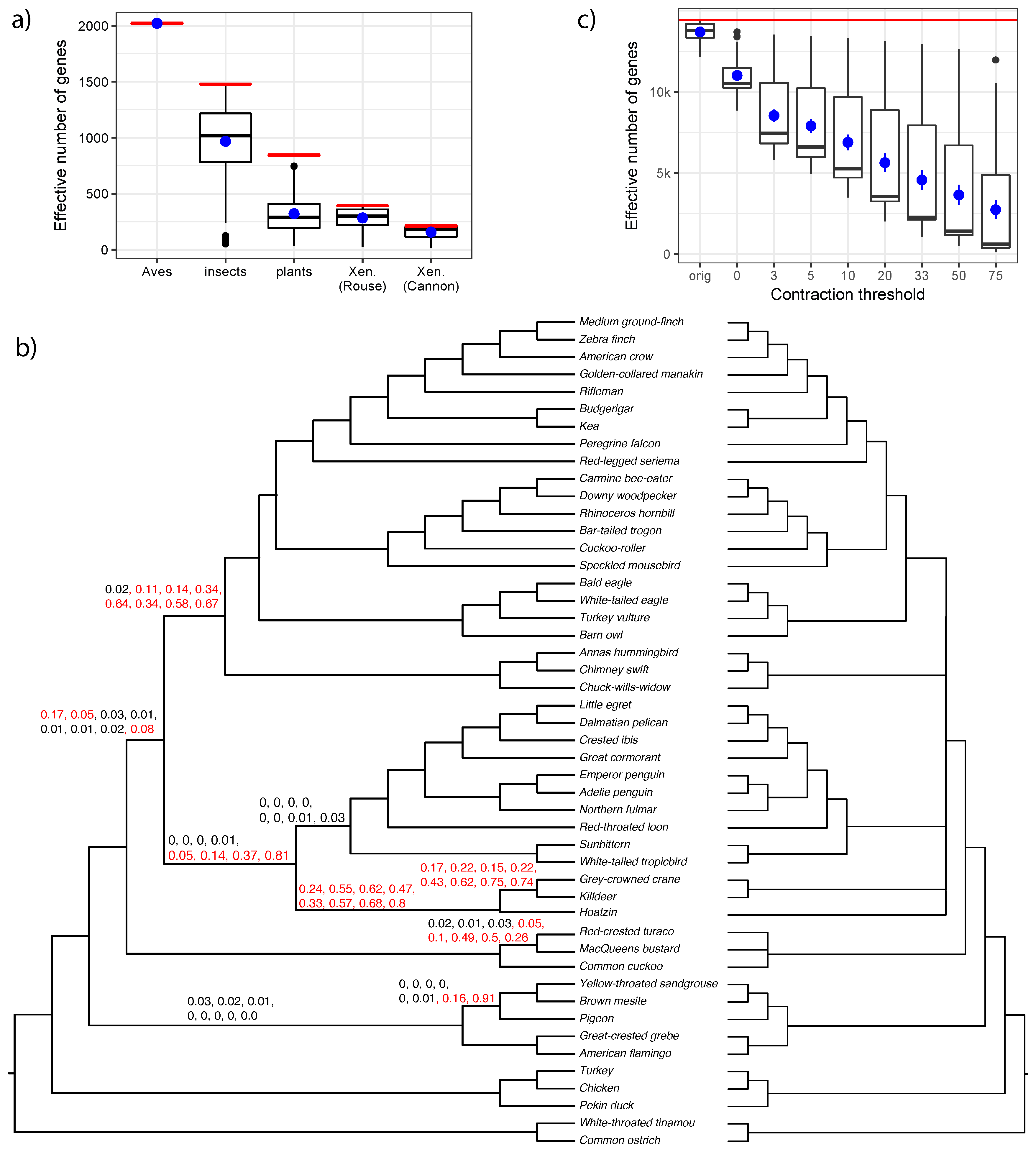
3.2. Biological Dataset
4. Discussion
4.1. Power
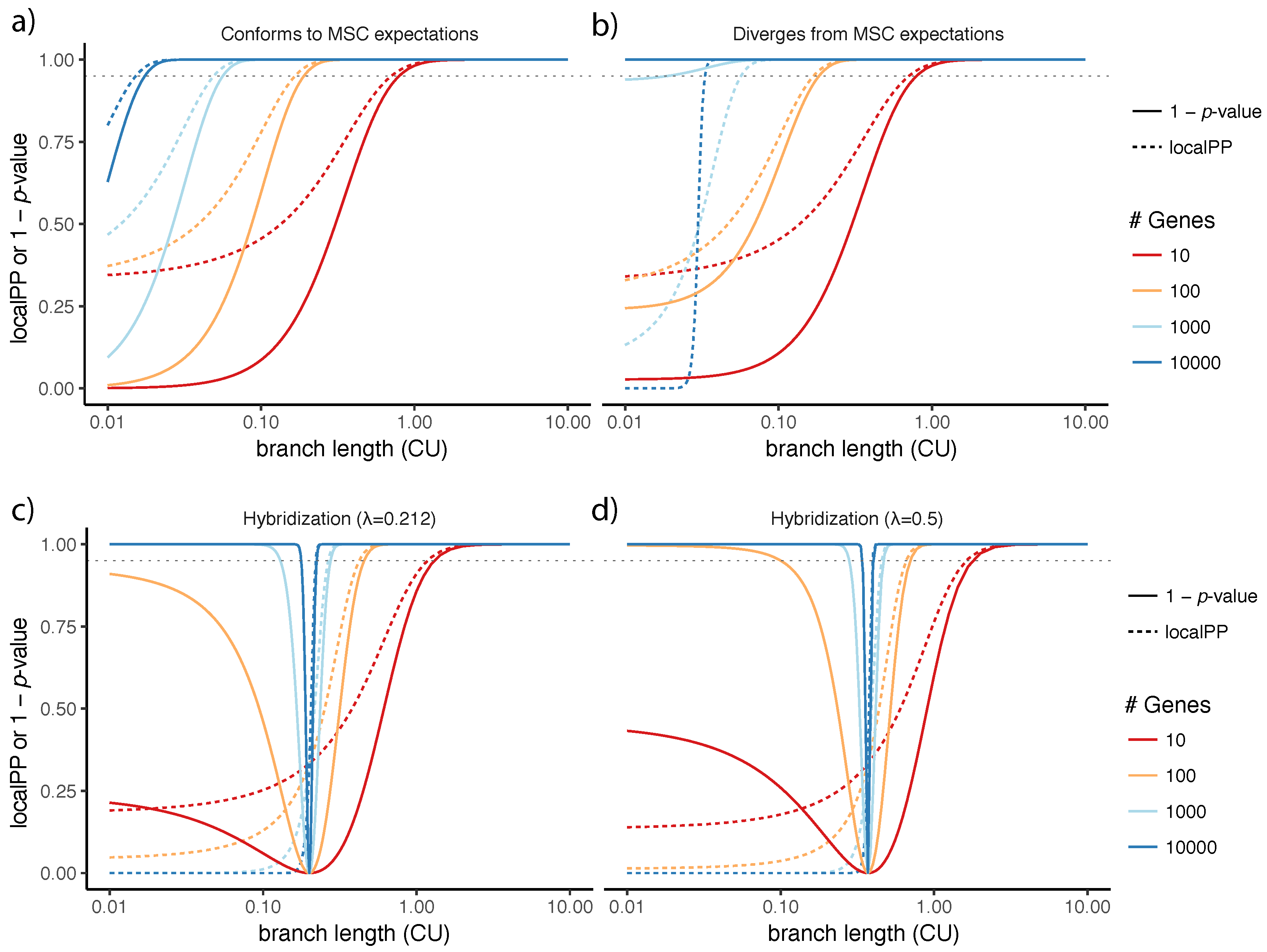
4.2. Divergence from the Multi-Species Coalescent Model and Connections to Local Posterior Probability
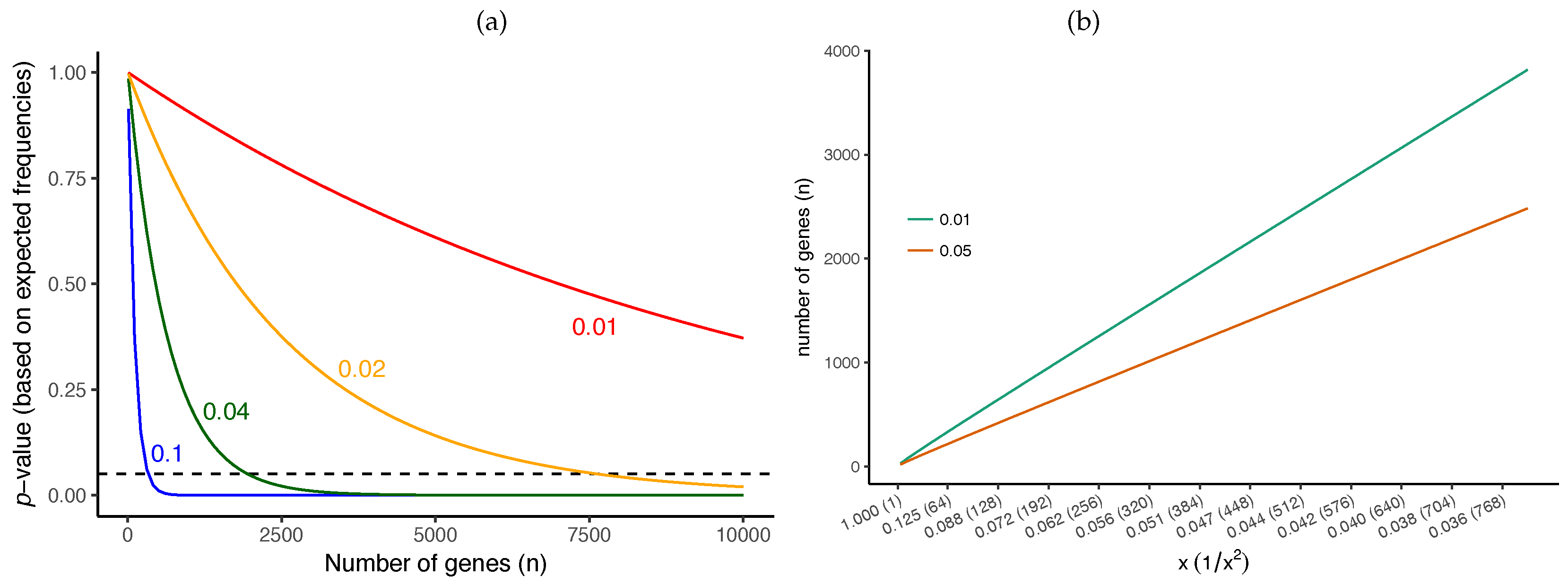
4.3. The Effective Number of Gene Trees
4.4. Interpretation
- Null hypothesis:
- The estimated gene tree quartets around the branch support all three NNI rearrangements around the branch in equal numbers.
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bapteste, E.; van Iersel, L.; Janke, A.; Kelchner, S.; Kelk, S.; McInerney, J.O.; Morrison, D.A.; Nakhleh, L.; Steel, M.; Stougie, L.; et al. Networks: Expanding evolutionary thinking. Trends Genet. 2013, 29, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Nakhleh, L. Evolutionary Phylogenetic Networks: Models and Issues. In Problem Solving Handbook in Computational Biology and Bioinformatics; Heath, L.S., Ramakrishnan, N., Eds.; Springer: Boston, MA, USA, 2011; pp. 125–158. [Google Scholar]
- Hoelzer, G.A.; Meinick, D.J. Patterns of speciation and limits to phylogenetic resolution. Trends Ecol. Evol. 1994, 9, 104–107. [Google Scholar] [CrossRef]
- Suh, A. The phylogenomic forest of bird trees contains a hard polytomy at the root of Neoaves. Zool. Scr. 2016, 45, 50–62. [Google Scholar] [CrossRef]
- Arntzen, J.W.; Themudo, G.E.; Wielstra, B. The phylogeny of crested newts (Triturus cristatus superspecies) nuclear and mitochondrial genetic characters suggest a hard polytomy, in line with the paleogeography of the centre of origin. Contrib. Zool. 2007, 76, 261–278. [Google Scholar]
- Townsend, J.P.; Su, Z.; Tekle, Y.I. Phylogenetic Signal and Noise: Predicting the Power of a Data Set to Resolve Phylogeny. Syst. Biol. 2012, 61, 835. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W. Reconstructing character evolution on polytomous cladograms. Cladistics 1989, 5, 365–377. [Google Scholar] [CrossRef]
- Chojnowski, J.L.; Kimball, R.T.; Braun, E.L. Introns outperform exons in analyses of basal avian phylogeny using clathrin heavy chain genes. Gene 2008, 410, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Jackman, T.R.; Larson, A.; de Queiroz, K.; Losos, J.B.; Cannatella, D. Phylogenetic Relationships and Tempo of Early Diversification in Anolis Lizards. Syst. Biol. 1999, 48, 254–285. [Google Scholar] [CrossRef]
- Walsh, H.E.; Kidd, M.G.; Moum, T.; Friesen, V.L. Polytomies and the power of phylogenetic inference. Evolution 1999, 53, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L.; Olsen, G.J.; Waddell, P.J.; Hillis, D.M. Phylogenetic inference. In Molecular Systematics, 2nd ed.; Hillis, D.M., Moritz, C., Mable, B.K., Eds.; Sinauer Associates, Inc.: Sunderland, MA, USA, 1996; pp. 407–514. [Google Scholar]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods); Version 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Braun, E.L.; Kimball, R.T. Polytomies, the power of phylogenetic inference, and the stochastic nature of molecular evolution: A comment on Walsh et al. (1999). Evolution 2001, 55, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O.; Sullivan, J. Approximate Likelihood-Ratio Test for Branches: A Fast, Accurate, and Powerful Alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.O.; Holder, M.T.; Holsinger, K.E. Polytomies and Bayesian phylogenetic inference. Syst. Biol. 2005, 54, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Slowinski, J.B. Molecular Polytomies. Mol. Phylogenet. Evol. 2001, 19, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P. Gene Trees in Species Trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Poe, S.; Chubb, A.L. Birds in a bush: Five genes indicate explosive evolution of avian orders. Evol. Int. J. Org. Evol. 2004, 58, 404–415. [Google Scholar] [CrossRef]
- Rannala, B.; Yang, Z. Bayes Estimation of Species Divergence Times and Ancestral Population Sizes Using DNA Sequences From Multiple Loci. Genetics 2003, 164, 1645–1656. [Google Scholar] [PubMed]
- Pamilo, P.; Nei, M. Relationships between gene trees and species trees. Mol. Biol. Evol. 1988, 5, 568–583. [Google Scholar] [PubMed]
- Heled, J.; Drummond, A.J. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 2010, 27, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. BEST: Bayesian estimation of species trees under the coalescent model. Bioinformatics 2008, 24, 2542–2543. [Google Scholar] [CrossRef] [PubMed]
- Chifman, J.; Kubatko, L.S. Quartet Inference from SNP Data Under the Coalescent Model. Bioinformatics 2014, 30, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.; Bouckaert, R.; Felsenstein, J.; Rosenberg, N.A.; Roychoudhury, A. Inferring species trees directly from biallelic genetic markers: Bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 2012, 29, 1917–1932. [Google Scholar] [CrossRef] [PubMed]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swenson, M.S.; Warnow, T. ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 2014, 30, i541–i548. [Google Scholar] [CrossRef] [PubMed]
- Mirarab, S.; Warnow, T. ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 2015, 31, i44–i52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sayyari, E.; Mirarab, S. ASTRAL-III: Increased Scalability and Impacts of Contracting Low Support Branches. In Comparative Genomics; Meidanis, J., Nakhleh, L., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 53–75. [Google Scholar]
- Larget, B.R.; Kotha, S.K.; Dewey, C.N.; Ané, C. BUCKy: Gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics 2010, 26, 2910–2911. [Google Scholar] [CrossRef] [PubMed]
- Sayyari, E.; Mirarab, S. Fast Coalescent-Based Computation of Local Branch Support from Quartet Frequencies. Mol. Biol. Evol. 2016, 33, 1654–1668. [Google Scholar] [CrossRef] [PubMed]
- Allman, E.S.; Degnan, J.H.; Rhodes, J.A. Identifying the rooted species tree from the distribution of unrooted gene trees under the coalescent. J. Math. Biol. 2011, 62, 833–862. [Google Scholar] [CrossRef] [PubMed]
- Zar, J.H. Biostatistical Analysis, 5th ed.; Prentice-Hall, Inc.: Upper Saddle River, NJ, USA, 2007. [Google Scholar]
- Koehler, K.J.; Larntz, K. An Empirical Investigation of Goodness-of-Fit Statistics for Sparse Multinomials. J. Am. Stat. Assoc. 1980, 75, 336–344. [Google Scholar] [CrossRef]
- Read, T.R.C.; Cressie, N.A.C. Goodness-of-Fit Statistics for Discrete Multivariate Data; Springer: New York, NY, USA, 1988. [Google Scholar]
- Hoschek, W. The Colt Distribution: Open Source Libraries for High Performance Scientific and Technical Computing in JAVA; CERN: Geneva, Switzerland, 2004. [Google Scholar]
- Sayyari, E.; Whitfield, J.B.; Mirarab, S. Fragmentary Gene Sequences Negatively Impact Gene Tree and Species Tree Reconstruction. Mol. Biol. Evol. 2017, 34, 3279–3291. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirarab, S.; Bayzid, M.S.; Boussau, B.; Warnow, T. Statistical binning enables an accurate coalescent-based estimation of the avian tree. Science 2014, 346, 1250463. [Google Scholar] [CrossRef] [PubMed]
- Rouse, G.W.; Wilson, N.G.; Carvajal, J.I.; Vrijenhoek, R.C. New deep-sea species of Xenoturbella and the position of Xenacoelomorpha. Nature 2016, 530, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.T.; Vellutini, B.C.; Smith, J.; Ronquist, F.; Jondelius, U.; Hejnol, A. Xenacoelomorpha is the sister group to Nephrozoa. Nature 2016, 530, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.J.; Matasci, N.; Ayyampalayam, S.; Barker, M.S.; Burleigh, J.G.; Gitzendanner, M.A.; et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 2014, 111, 4859–4868. [Google Scholar] [CrossRef] [PubMed]
- Mallo, D.; De Oliveira Martins, L.; Posada, D. SimPhy: Phylogenomic Simulation of Gene, Locus, and Species Trees. Syst. Biol. 2016, 65, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, W.; Yang, Z. INDELible: A flexible simulator of biological sequence evolution. Mol. Biol. Evol. 2009, 26, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Degnan, J.H.; Rosenberg, N.A. Discordance of Species Trees with Their Most Likely Gene Trees. PLoS Genet. 2006, 2, e68. [Google Scholar] [CrossRef] [PubMed]
- Degnan, J.H. Anomalous Unrooted Gene Trees. Syst. Biol. 2013, 62, 574–590. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.; Buchanan, K.L. A quantum leap in avian biology. Emu 2015, 115, 1–5. [Google Scholar] [CrossRef]
- Swati Patel, R.T.K.; Braun, E.L. Error in Phylogenetic Estimation for Bushes in the Tree of Life. J. Phylogenet. Evol. Biol. 2013, 1, 1–10. [Google Scholar]
- Shekhar, S.; Roch, S.; Mirarab, S. Species tree estimation using ASTRAL: How many genes are enough? IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Erdos, P.; Steel, M.; Szekely, L.; Warnow, T. A few logs suffice to build (almost) all trees: Part II. Theor. Comput. Sci. 1999, 221, 77–118. [Google Scholar] [CrossRef]
- Solís-Lemus, C.; Yang, M.; Ané, C. Inconsistency of Species Tree Methods under Gene Flow. Syst. Biol. 2016, 65, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.S.; Gatesy, J. On the importance of homology in the age of phylogenomics. Syst. Biodivers. 2017, 1–19. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Anderson, D.R.; Burnham, K.P.; Thompson, W.L. Null Hypothesis Testing: Problems, Prevalence, and an Alternative. J. Wildl. Manag. 2000, 64, 912–923. [Google Scholar] [CrossRef]
- Goodman, S. A Dirty Dozen: Twelve p-Value Misconceptions. Semin. Hematol. 2008, 45, 135–140. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Dataset | Ref | Max Height | N | n | R | ||
|---|---|---|---|---|---|---|---|---|
| Biological | Aves | [37] | 48 | 2022 | 1 | 0.64 | ||
| insect | [36] | 144 | 1478 | 1 | 0.72 | |||
| plant | [41] | 103 | 844 | 1 | 0.89 | |||
| Xenoturbella | [39] | 26 | 393 | 1 | 0.50 | |||
| Xenoturbella | [40] | 78 | 212 | 1 | 0.55 | |||
| Simulated | S12A | new | 1.6 M | 12 | 1000 | 50 | 0.82 | 36% |
| S12B | new | 1.6 M | 12 | 1000 | 50 | 0.68 | 35% | |
| S201 | [27] | 10 M | 201 | 1000 | 100 | 0.94 | 25% | |
| S201 | [27] | 2 M | 201 | 1000 | 100 | 0.72 | 31% | |
| S201 | [27] | 500 K | 201 | 1000 | 97 | 0.49 | 47% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayyari, E.; Mirarab, S. Testing for Polytomies in Phylogenetic Species Trees Using Quartet Frequencies. Genes 2018, 9, 132. https://doi.org/10.3390/genes9030132
Sayyari E, Mirarab S. Testing for Polytomies in Phylogenetic Species Trees Using Quartet Frequencies. Genes. 2018; 9(3):132. https://doi.org/10.3390/genes9030132
Chicago/Turabian StyleSayyari, Erfan, and Siavash Mirarab. 2018. "Testing for Polytomies in Phylogenetic Species Trees Using Quartet Frequencies" Genes 9, no. 3: 132. https://doi.org/10.3390/genes9030132
APA StyleSayyari, E., & Mirarab, S. (2018). Testing for Polytomies in Phylogenetic Species Trees Using Quartet Frequencies. Genes, 9(3), 132. https://doi.org/10.3390/genes9030132