The Role of microRNAs in Alzheimer’s Disease and Their Therapeutic Potentials


 ,
,  ,
,
Abstract
:1. Introduction
1.1. Amyloidogenesis
1.2. Tauopathy
1.3. Neuroinflammation
1.4. Synaptic Dysfunction
2. Current Drug Therapies for Alzheimer’s Disease
2.1. BACE1 Inhibitors
2.2. γ-Secretase Inhibitors and Modulators
2.3. Aβ Immunotherapy
2.4. Acetylcholinesterase Inhibitors
2.5. NMDA Receptor Antagonists
3. miRNAs in Alzheimer’s Disease
3.1. Rationale for miRNA Investigation in AD
3.2. miRNA Biogenesis and Function
3.3. miRNA Dysregulation in Alzheimer’s Disease
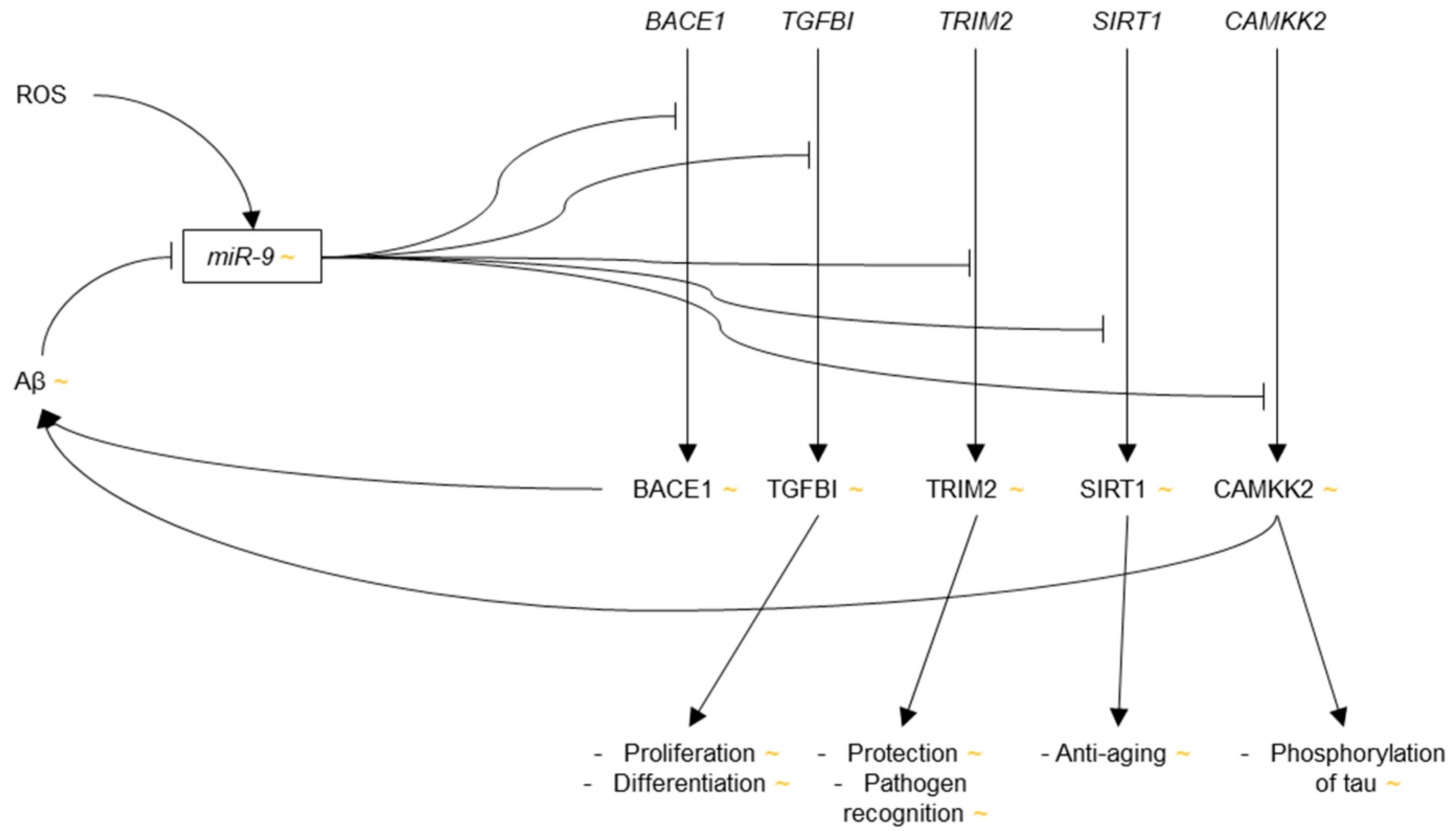
3.3.1. microRNA-9
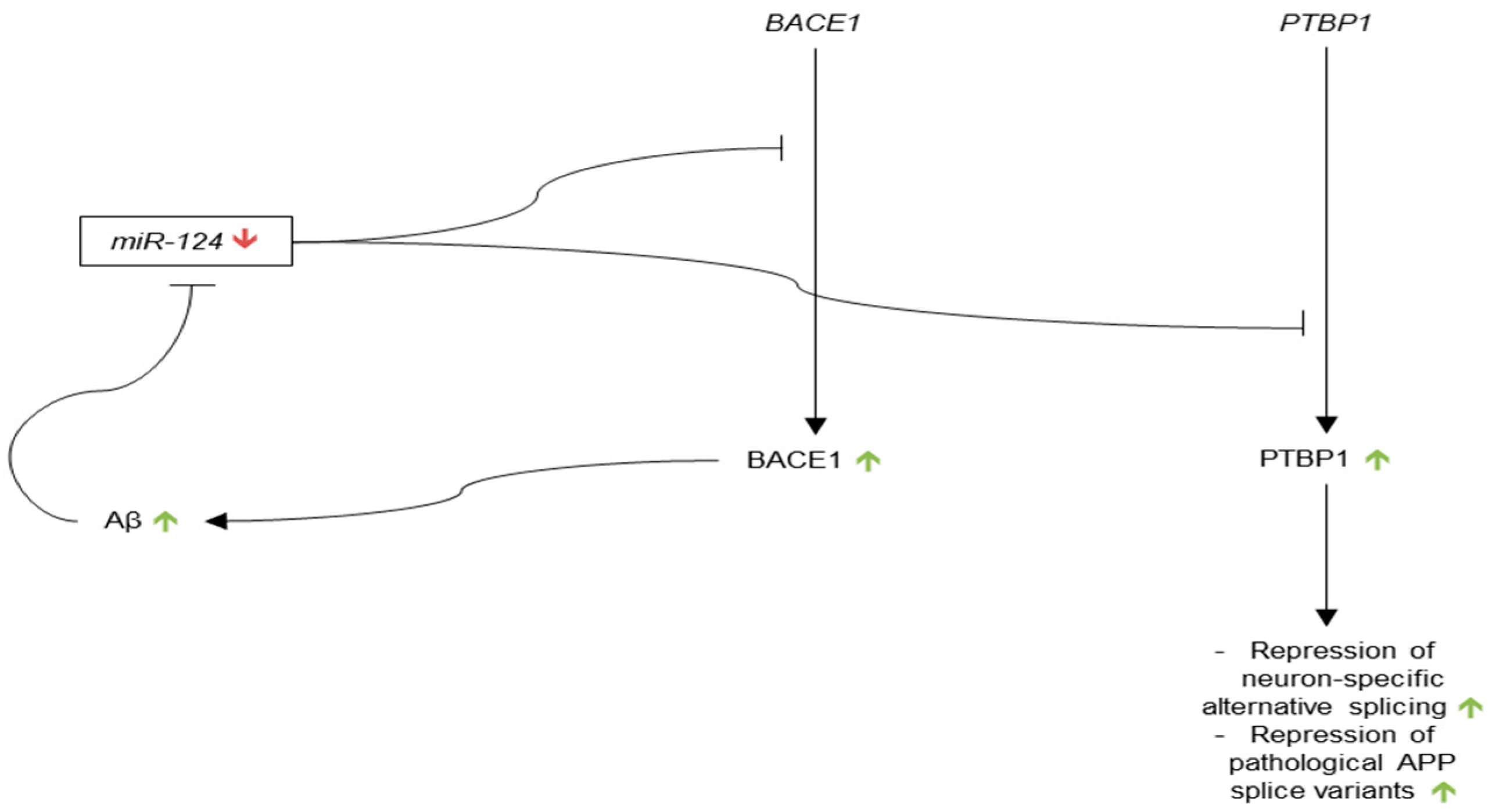
3.3.2. microRNA-124
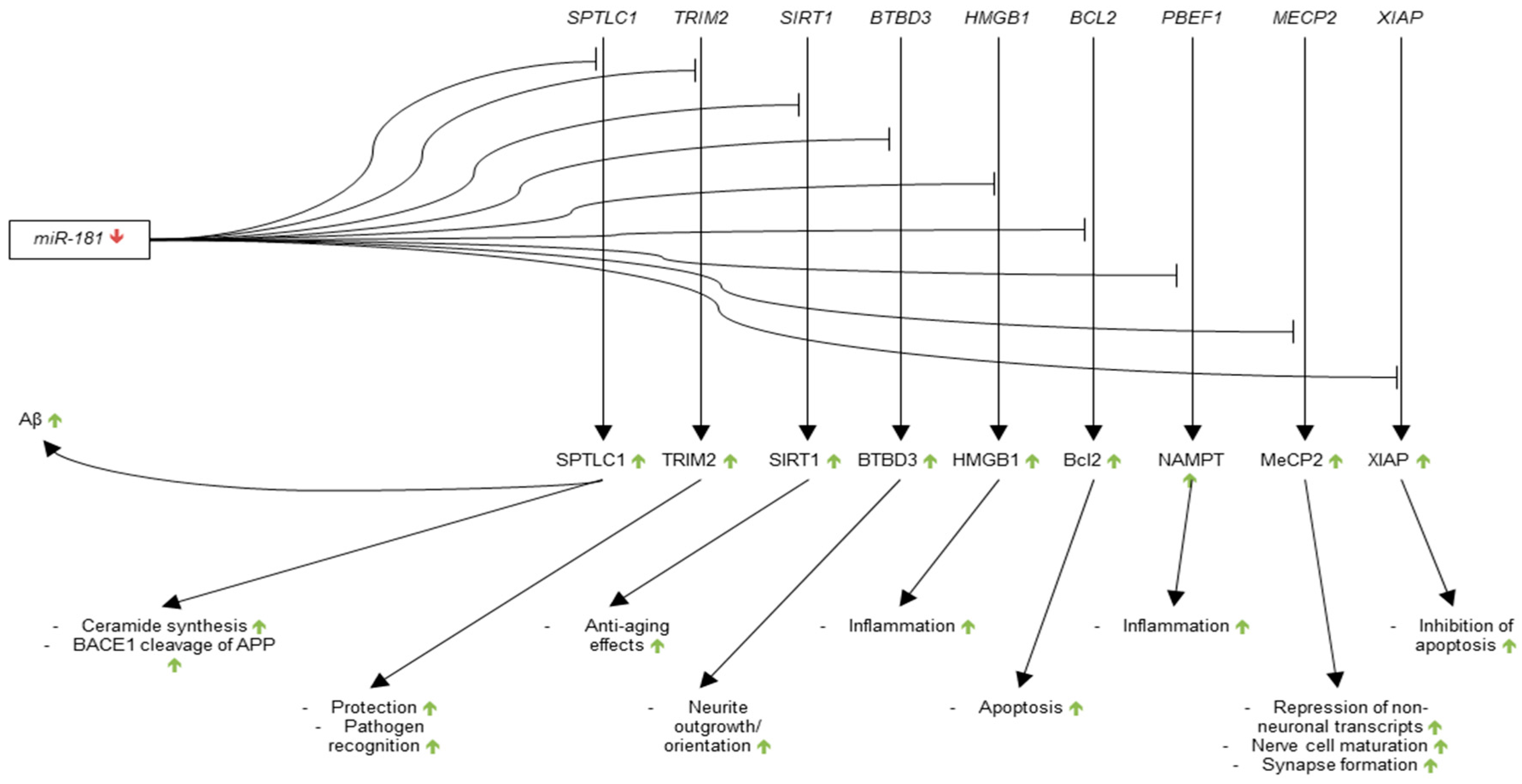
3.3.3. microRNA-181
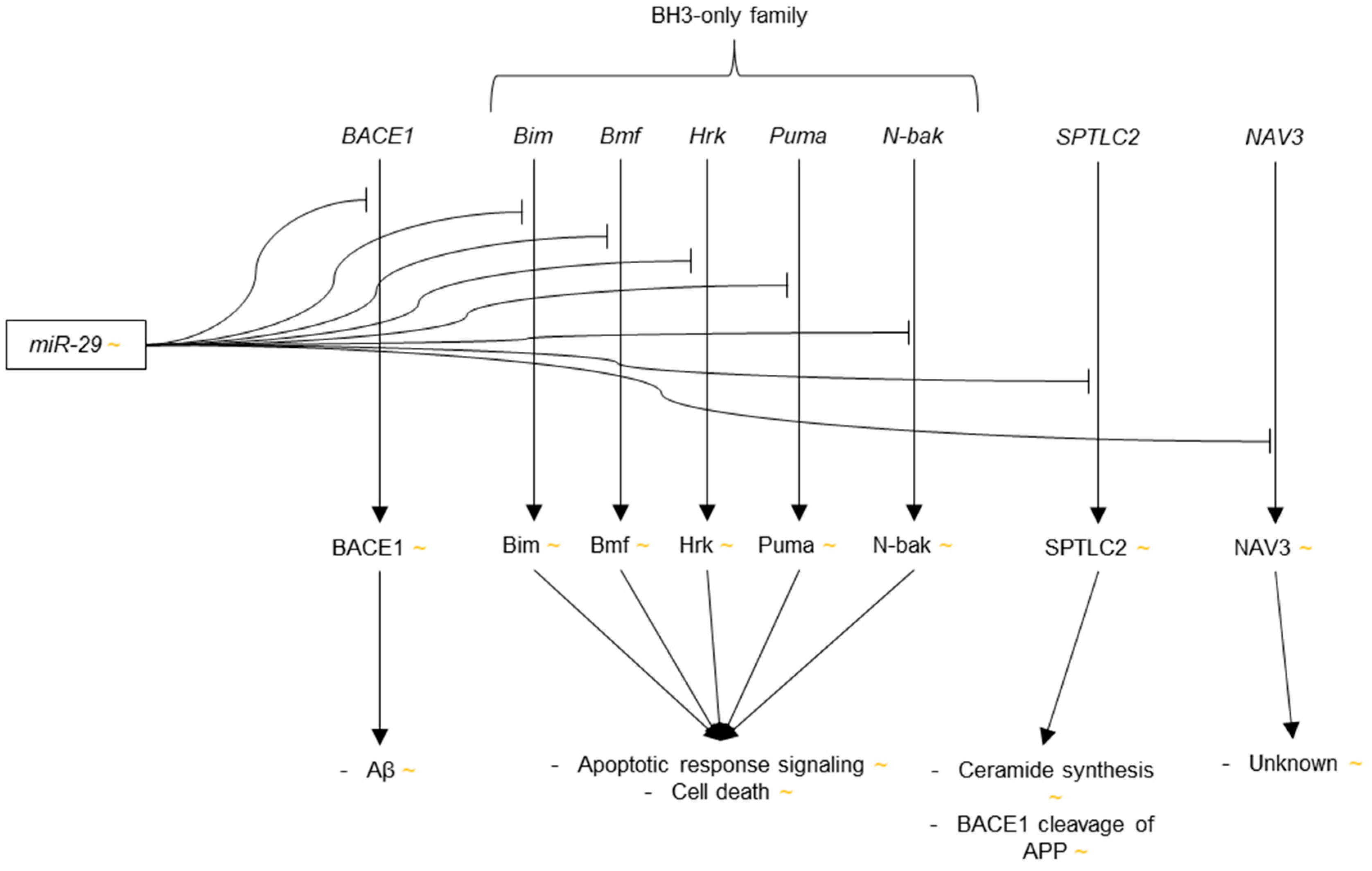
3.3.4. microRNA-29
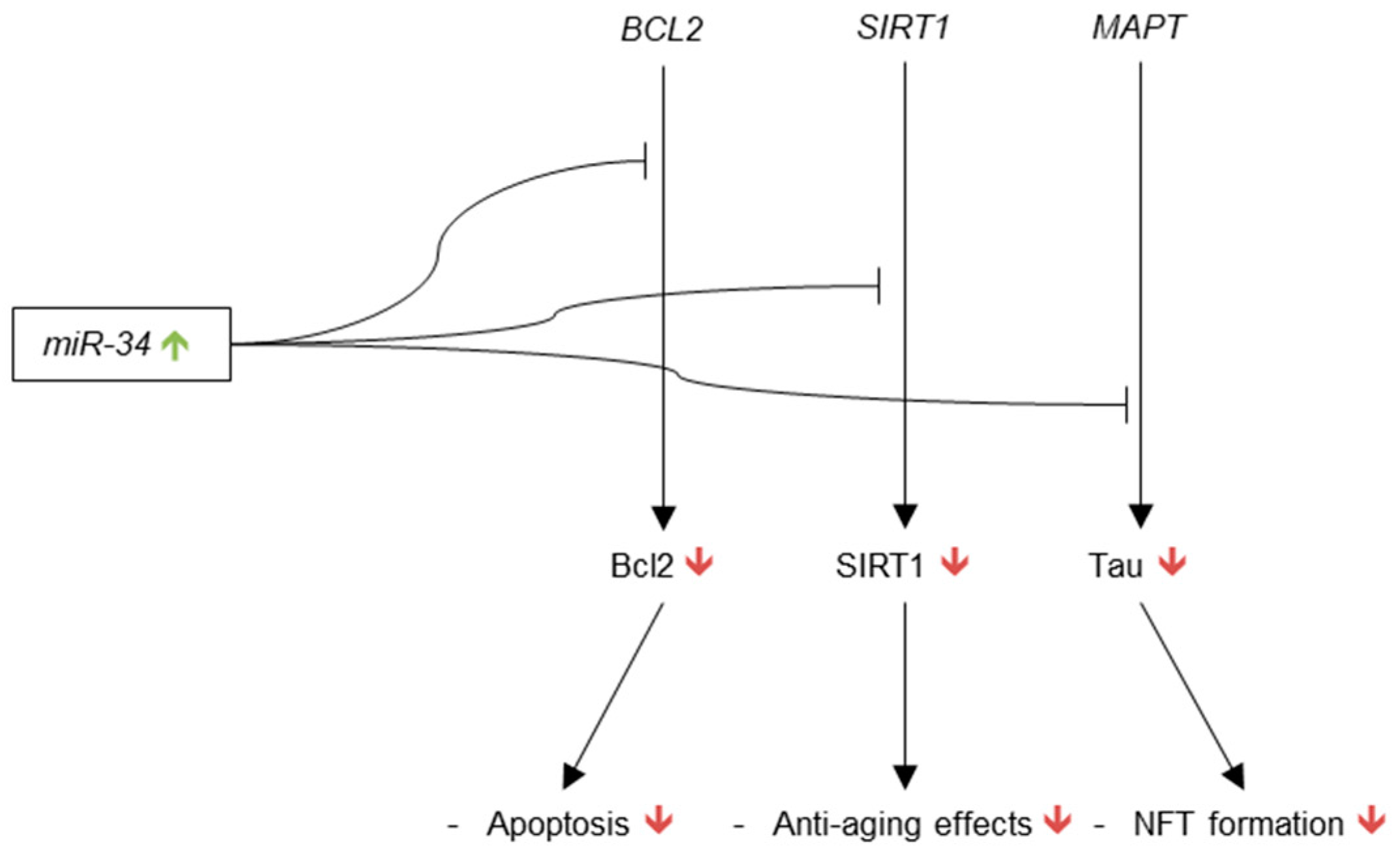
3.3.5. microRNA-34
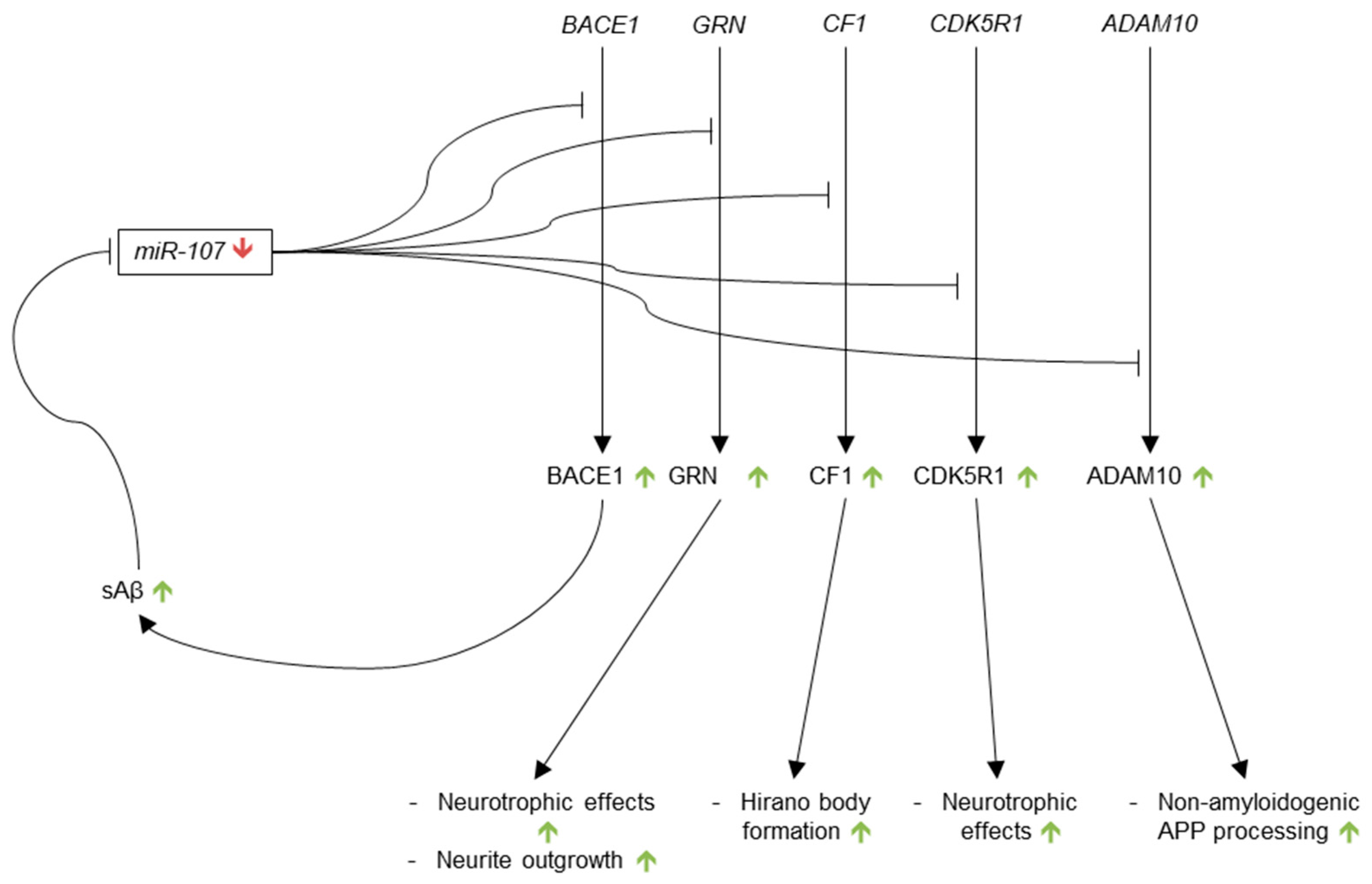
3.3.6. microRNA-107
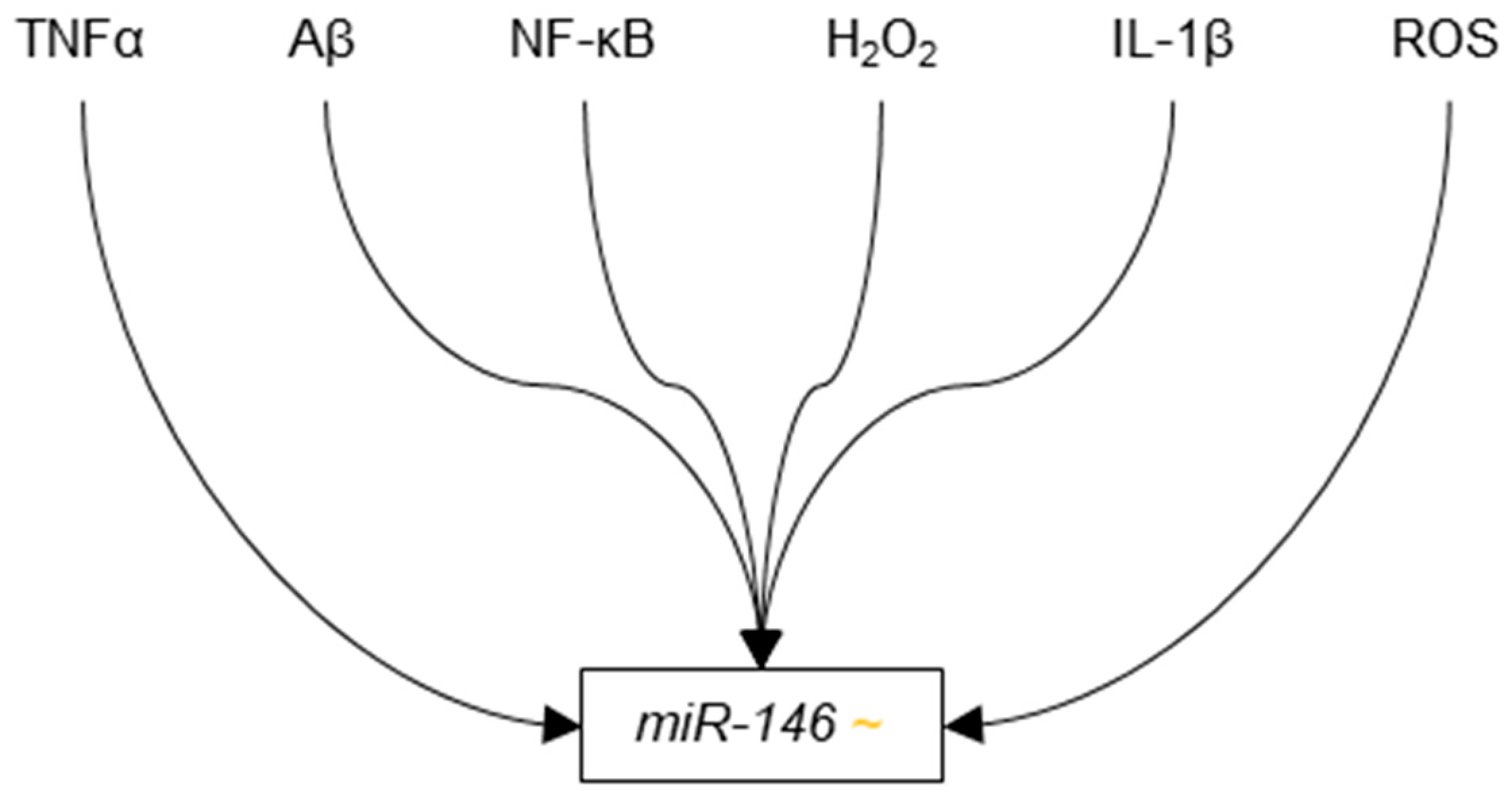
3.3.7. microRNA-146
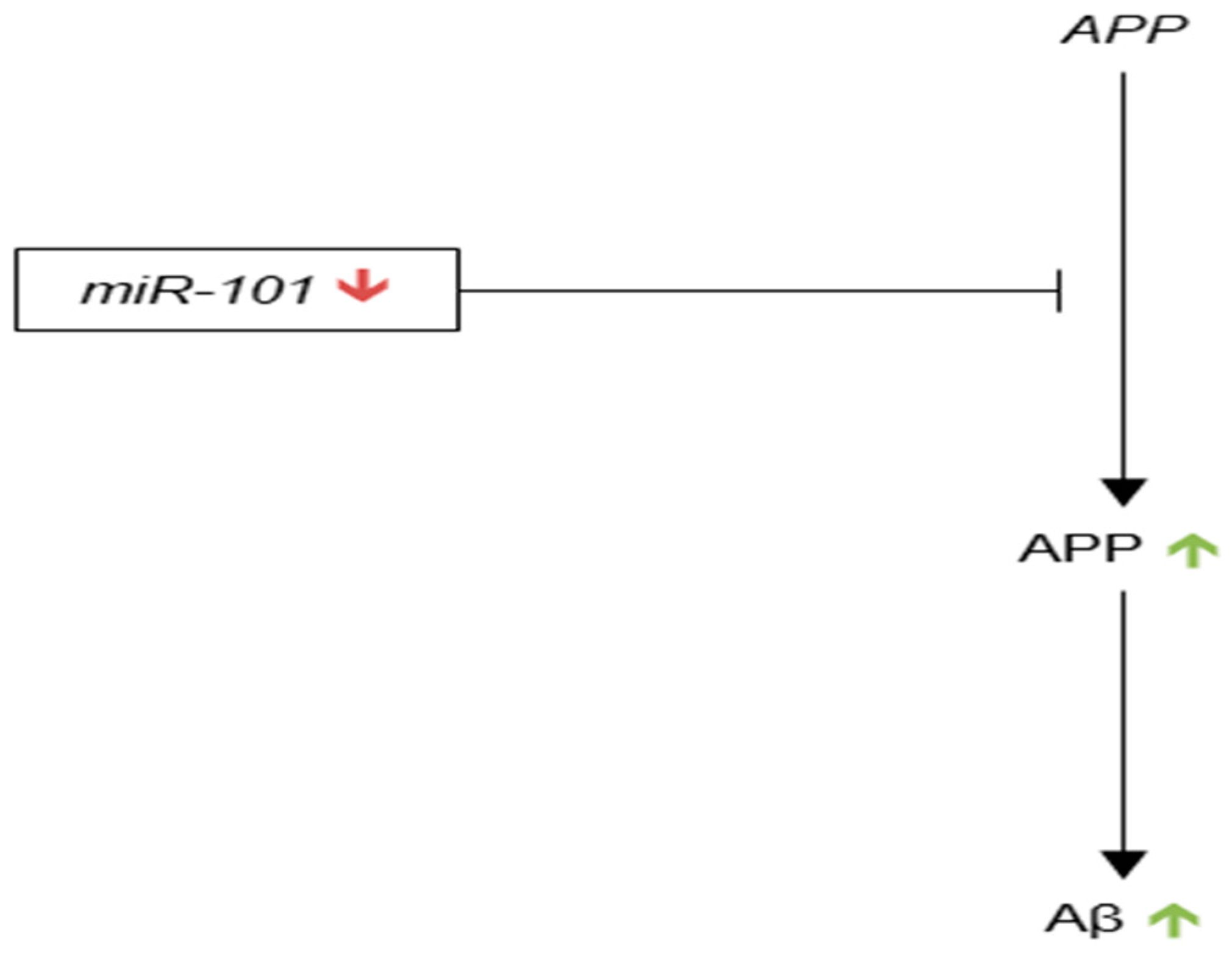
3.3.8. microRNA-101
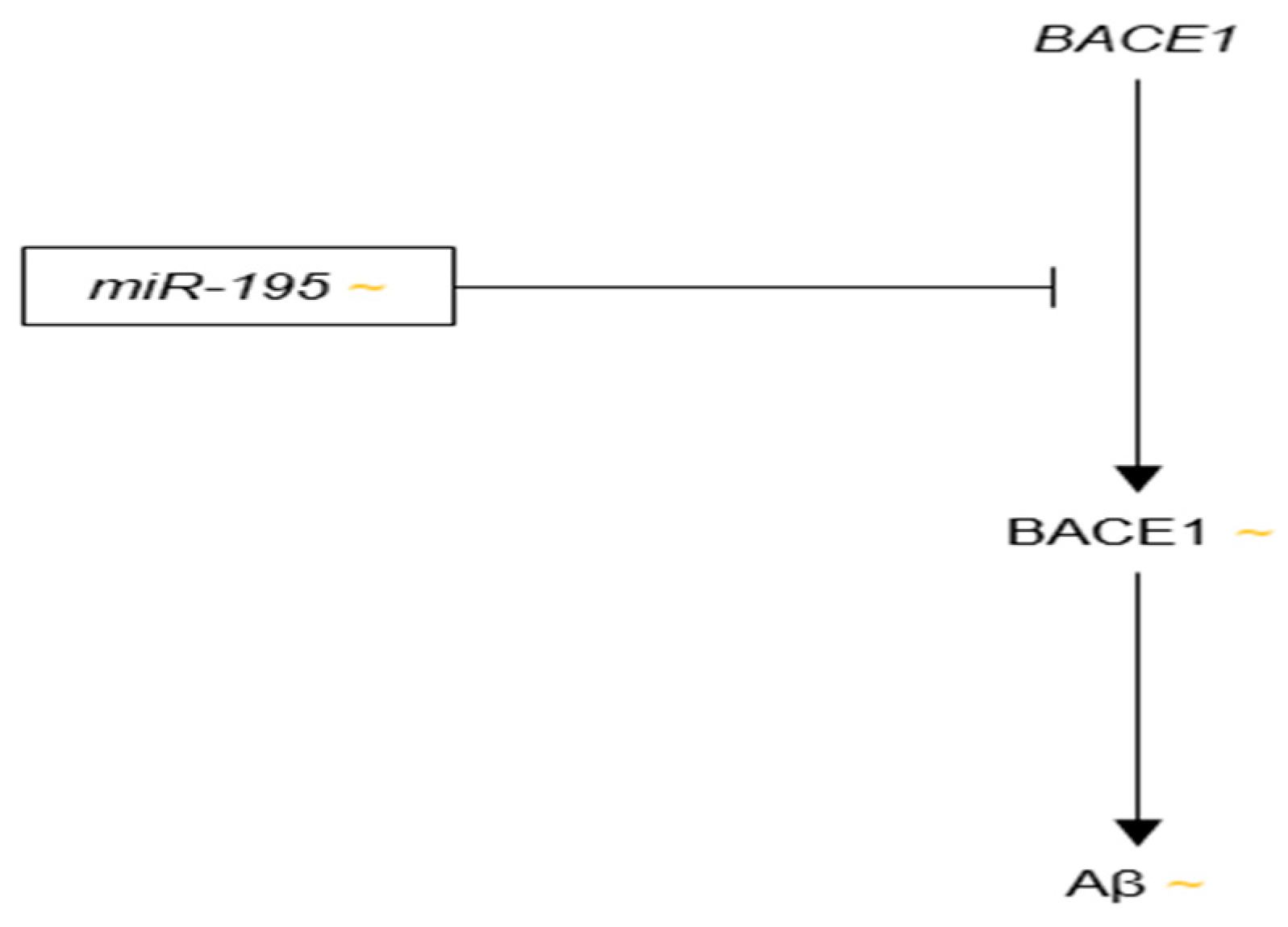
3.3.9. microRNA-195
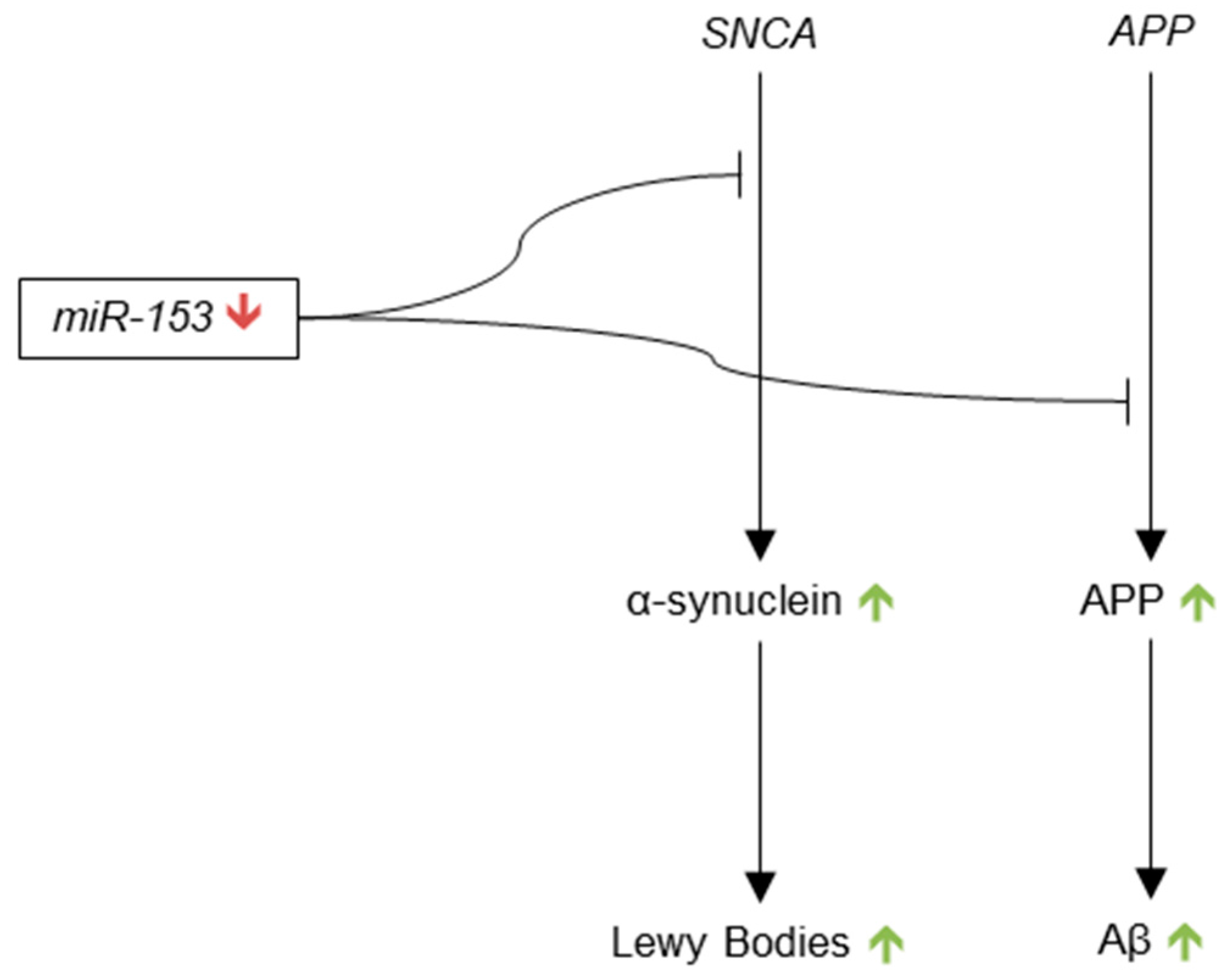
3.3.10. microRNA-153
3.3.11. microRNA-132/212
4. Therapeutic Targets for miRNAs
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jellinger, K.A. Clinicopathological analysis of dementia disorders in the elderly—An update. J. Alzheimers Dis. 2006, 9, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.; Danielczyk, W.; Fischer, P.; Gabriel, E. Clinicopathological analysis of dementia disorders in the elderly. J. Neurol. Sci. 1990, 95, 239–258. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the national institute on aging-alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Albert, M.S.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carrillo, M.C.; Thies, B.; Phelps, C.H. Introduction to the recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for alzheimer’s disease. Alzheimers Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of alzheimer’s disease: Recommendations from the national institute on aging-alzheimer’s association workgroups on diagnostic guidelines for alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the national institute on aging-alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Association, A.S. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014, 10, e47–e92. [Google Scholar]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J. Association of apolipoprotein E allele ε 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; Huang, Y.; Ashford, J.W. Apoe genotype accounts for the vast majority of ad risk and ad pathology. Neurobiol. Aging 2004, 25, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.; Kivipelto, M.; Wolozin, B.; Zhou, J.; Whitmer, R.A. Midlife serum cholesterol and increased risk of alzheimer’s and vascular dementia three decades later. Dement. Geriatr. Cogn. Disord. 2009, 28, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Launer, L.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.; Foley, D.; White, L.R.; Havlik, R.J. Midlife blood pressure and dementia: The honolulu-asia aging study. Neurobiol. Aging 2000, 21, 49–55. [Google Scholar] [CrossRef]
- Ahtiluoto, S.; Polvikoski, T.; Peltonen, M.; Solomon, A.; Tuomilehto, J.; Winblad, B.; Sulkava, R.; Kivipelto, M. Diabetes, alzheimer disease, and vascular dementia: A population-based neuropathologic study. Neurology 2010, 75, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Noble, J.; Tang, M.X.; Schupf, N.; Mayeux, R.; Luchsinger, J.A. Type 2 diabetes and late-onset Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2011, 31, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64 (Suppl. 9), 7–10. [Google Scholar] [PubMed]
- Willem, M.; Garratt, A.N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C. Control of peripheral nerve myelination by the β-secretase BACE1. Science 2006, 314, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.W. The γ-secretase complex: From structure to function. Front. Cell. Neurosci. 2014, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.; Bieger, S.C.; Brühl, B.; Tienari, P.J.; Ida, N.; Allsop, D.; Roberts, G.W.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; et al. Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides. Nat. Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.G.; Forman, M.S.; Sung, J.C.; Leight, S.; Kolson, D.L.; Iwatsubo, T.; Lee, V.M.; Doms, R.W. Alzheimer’s A beta(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat. Med. 1997, 3, 1021–1023. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, J.P.; Tsai, J.; Gouras, G.K.; Hai, B.; Thinakaran, G.; Checler, F.; Sisodia, S.S.; Greengard, P.; Xu, H. Endoplasmic reticulum and trans-golgi network generate distinct populations of alzheimer β-amyloid peptides. Proc. Natl. Acad. Sci. USA 1999, 96, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Asai, M.; Hattori, C.; Szabó, B.; Sasagawa, N.; Maruyama, K.; Tanuma, S.; Ishiura, S. Putative function of ADAM9, ADAM10, and ADAM17 as APP α-secretase. Biochem. Biophys. Res. Commun. 2003, 301, 231–235. [Google Scholar] [CrossRef]
- Tanabe, C.; Hotoda, N.; Sasagawa, N.; Sehara-Fujisawa, A.; Maruyama, K.; Ishiura, S. ADAM19 is tightly associated with constitutive Alzheimer’s disease app α-secretase in a172 cells. Biochem. Biophys. Res. Commun. 2007, 352, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Igbavboa, U.; Eckert, G.P.; Malo, T.M.; Studniski, A.E.; Johnson, L.N.; Yamamoto, N.; Kobayashi, M.; Fujita, S.C.; Appel, T.R.; Müller, W.E.; et al. Murine synaptosomal lipid raft protein and lipid composition are altered by expression of human apoE3 and 4 and by increasing age. J. Neurol. Sci. 2005, 229–230, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gamache, E.; Rosenman, D.J.; Xie, J.; Lopez, M.M.; Li, Y.M.; Wang, C. Familial Alzheimer’s mutations within APPtm increase aβ42 production by enhancing accessibility of ε-cleavage site. Nat. Commun. 2014, 5, 3037. [Google Scholar] [CrossRef] [PubMed]
- Lemere, C.A.; Lopera, F.; Kosik, K.S.; Lendon, C.L.; Ossa, J.; Saido, T.C.; Yamaguchi, H.; Ruiz, A.; Martinez, A.; Madrigal, L.; et al. The E280A presenilin 1 Alzheimer mutation produces increased Aβ 42 deposition and severe cerebellar pathology. Nat. Med. 1996, 2, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.S.; Martinez, M.; Brunkan, A.L.; Goate, A. Presenilin 2 familial Alzheimer’s disease mutations result in partial loss of function and dramatic changes in Aβ 42/40 ratios. J. Neurochem. 2005, 92, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Spies, P.E.; Slats, D.; Sjögren, J.M.; Kremer, B.P.; Verhey, F.R.; Rikkert, M.G.; Verbeek, M.M. The cerebrospinal fluid amyloid-β 42/40 ratio in the differentiation of Alzheimer’s disease from non-alzheimer’s dementia. Curr. Alzheimer Res. 2010, 7, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by alzheimer’s-related amyloid β oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The relationship between amyloid and tau. J. Mol. Neurosci. 2003, 20, 203–206. [Google Scholar] [CrossRef]
- Lloret, A.; Fuchsberger, T.; Giraldo, E.; Viña, J. Molecular mechanisms linking amyloid β toxicity and tau hyperphosphorylation in alzheimer’s disease. Free Radic. Biol. Med. 2015, 83, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Hartlage-Rübsamen, M.; Zeitschel, U.; Apelt, J.; Gärtner, U.; Franke, H.; Stahl, T.; Günther, A.; Schliebs, R.; Penkowa, M.; Bigl, V.; et al. Astrocytic expression of the Alzheimer’s disease β-secretase (BACE1) is stimulus-dependent. Glia 2003, 41, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Weggen, S.; Diehlmann, A.; Buslei, R.; Beyreuther, K.; Bayer, T.A. Prominent expression of presenilin-1 in senile plaques and reactive astrocytes in Alzheimer’s disease brain. Neuroreport 1998, 9, 3279–3283. [Google Scholar] [PubMed]
- Davies, C.A.; Mann, D.M.; Sumpter, P.Q.; Yates, P.O. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J. Neurol. Sci. 1987, 78, 151–164. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Geinisman, Y.; de Toledo-Morrell, L.; Morrell, F. Aged rats need a preserved complement of perforated axospinous synapses per hippocampal neuron to maintain good spatial memory. Brain Res. 1986, 398, 266–275. [Google Scholar] [CrossRef]
- Geinisman, Y.; de Toledo-Morrell, L.; Morrell, F. Loss of perforated synapses in the dentate gyrus: Morphological substrate of memory deficit in aged rats. Proc. Natl. Acad. Sci. USA 1986, 83, 3027–3031. [Google Scholar] [CrossRef] [PubMed]
- Geinisman, Y.; deToledo-Morrell, L.; Morrell, F.; Persina, I.S.; Rossi, M. Age-related loss of axospinous synapses formed by two afferent systems in the rat dentate gyrus as revealed by the unbiased stereological dissector technique. Hippocampus 1992, 2, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Rapp, P.R.; Gallagher, M. Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc. Natl. Acad. Sci. USA 1996, 93, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.; Schliemann, T.; Sørensen, J.C.; Zimmer, J.; West, M.J. Memory impaired aged rats: No loss of principal hippocampal and subicular neurons. Neurobiol. Aging 1996, 17, 143–147. [Google Scholar] [CrossRef]
- Sze, C.I.; Troncoso, J.C.; Kawas, C.; Mouton, P.; Price, D.L.; Martin, L.J. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Mani, G.; Park, B.S.; Jacques, J.; Murdoch, G.; Whetsell, W.; Kaye, J.; Manczak, M. Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimers Dis. 2005, 7, 103–117, discussion 173–180. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and Aβ. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Townsend, M.; Podlisny, M.B.; Shankar, G.M.; Fadeeva, J.V.; El Agnaf, O.; Hartley, D.M.; Selkoe, D.J. Certain inhibitors of synthetic amyloid β-peptide (Aβ) fibrillogenesis block oligomerization of natural Aβ and thereby rescue long-term potentiation. J. Neurosci. 2005, 25, 2455–2462. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [PubMed]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tsien, J.Z. Memory and the NMDA receptors. N. Engl. J. Med. 2009, 361, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. Ampar removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. β-amyloid1–42 binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef] [PubMed]
- Garzon, D.J.; Fahnestock, M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J. Neurosci. 2007, 27, 2628–2635. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yan, R. Inhibition of BACE1 for therapeutic use in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2010, 3, 618–628. [Google Scholar] [PubMed]
- Strobel, G. Keystone Drug News: Comentis BACE Inhibitor Debuts. Available online: https://www.alzforum.org/news/conference-coverage/keystone-drug-news-comentis-bace-inhibitor-debuts (accessed on 8 January 2018).
- Forman, M.; Palcza, J.; Tseng, J.; Leempoels, J.; Ramael, S.; Han, D.; Jhee, S.; Ereshefsky, L.; Tanen, M.; Laterza, O.; et al. The novel bace inhibitor MK-8931 dramatically lowers cerebrospinal fluid Aβ peptides in healthy subjects following single- and multiple-dose administration. In Proceedings of the 64th American Academy of Neurology Annual Meeting, New Orleans, LA, USA, 21–28 April 2012. [Google Scholar]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; et al. The potent BACE1 inhibitor ly2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Menting, K.W.; Claassen, J.A. β-secretase inhibitor; a promising novel therapeutic drug in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Chavez Gutierrez, L. Learning by failing: Ideas and concepts to tackle γ-secretases in Alzheimer’s disease and beyond. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Veugelen, S.; Dewilde, M.; De Strooper, B.; Chavez-Gutierrez, L. Screening and characterization strategies for nanobodies targeting membrane proteins. Methods Enzymol. 2017, 584, 59–97. [Google Scholar] [PubMed]
- Wisniewski, T.; Goni, F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron 2015, 85, 1162–1176. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical effects of Aβ immunization (an1792) in patients with ad in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604. [Google Scholar] [CrossRef]
- Ryan, J.M.; Grundman, M. Anti-amyloid-β immunotherapy in Alzheimer’s disease: Acc-001 clinical trials are ongoing. J. Alzheimers Dis. 2009, 17, 243. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.; Sano, M. Alzheimer disease: Solanezumab—Prospects for meaningful interventions in AD? Nat. Rev. Neurol. 2015, 11, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Ehret, M.J.; Chamberlin, K.W. Current practices in the treatment of alzheimer disease: Where is the evidence after the phase III trials? Clin. Ther. 2015, 37, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Alexandrov, P.N.; Lukiw, W.J. Anti-microRNAs as novel therapeutic agents in the clinical management of Alzheimer’s disease. Front. Neurosci. 2016, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Melo, C.A.; Melo, S.A. Biogenesis and physiology of microRNAs. In Non-Coding Rnas and Cancer; Springer: New York, NY, USA, 2014; pp. 5–24. [Google Scholar]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E.; Guryev, V.; van de Belt, J.; Wienholds, E.; Plasterk, R.H.; Cuppen, E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell 2005, 120, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Holohan, K.N.; Lahiri, D.K.; Schneider, B.P.; Foroud, T.; Saykin, A.J. Functional microRNAs in Alzheimer’s disease and cancer: Differential regulation of common mechanisms and pathways. Front. Genet. 2012, 3, 323. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.K.; Cheung, T.H.; Huen, N.Y.; Wong, K.W.; Lo, K.W.; Yim, S.F.; Siu, N.S.; Wong, Y.M.; Tsang, P.T.; Pang, M.W.; et al. Dysregulated microRNAs and their predicted targets associated with endometrioid endometrial adenocarcinoma in Hong Kong women. Int. J. Cancer 2009, 124, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yu, J.T.; Tan, M.S.; Liu, Q.Y.; Wang, H.F.; Zhang, W.; Jiang, T.; Tan, L. Genome-wide serum microRNA expression profiling identifies serum biomarkers for Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 1017–1027. [Google Scholar] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Sempere, L.F.; Freemantle, S.; Pitha-Rowe, I.; Moss, E.; Dmitrovsky, E.; Ambros, V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004, 5, R13. [Google Scholar] [CrossRef] [PubMed]
- Bak, M.; Silahtaroglu, A.; Møller, M.; Christensen, M.; Rath, M.F.; Skryabin, B.; Tommerup, N.; Kauppinen, S. MicroRNA expression in the adult mouse central nervous system. RNA 2008, 14, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 2007, 18, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Pogue, A.I. Induction of specific micro RNA (miRNA) species by ROS-generating metal sulfates in primary human brain cells. J. Inorg. Biochem. 2007, 101, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster mir-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/β-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [PubMed]
- Sethi, P.; Lukiw, W.J. Micro-RNA abundance and stability in human brain: Specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009, 459, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Schonrock, N.; Ke, Y.D.; Humphreys, D.; Staufenbiel, M.; Ittner, L.M.; Preiss, T.; Götz, J. Neuronal microRNA deregulation in response to Alzheimer’s disease amyloid-β. PLoS ONE 2010, 5, e11070. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Huang, Q.; Hu, Y.; Stromberg, A.J.; Nelson, P.T. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: White matter versus gray matter. Acta Neuropathol. 2011, 121, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Alexandrov, P.N. Regulation of complement factor h (CFH) by multiple miRNAs in Alzheimer’s disease (AD) brain. Mol. Neurobiol. 2012, 46, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ylikallio, E.; Pöyhönen, R.; Zimon, M.; De Vriendt, E.; Hilander, T.; Paetau, A.; Jordanova, A.; Lönnqvist, T.; Tyynismaa, H. Deficiency of the E3 ubiquitin ligase TRIM2 in early-onset axonal neuropathy. Hum. Mol. Genet. 2013, 22, 2975–2983. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal sirt1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [PubMed]
- Schonrock, N.; Humphreys, D.T.; Preiss, T.; Gotz, J. Target gene repression mediated by miRNAs mir-181c and mir-9 both of which are down-regulated by amyloid-β. J. Mol. Neurosci. 2012, 46, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Zhang, L.H.; Xu, W.P.; Jing, P.; Zhan, P.Y. MicroRNA-9 attenuates amyloidβ-induced synaptotoxicity by targeting calcium/calmodulin-dependent protein kinase kinase 2. Mol. Med. Rep. 2014, 9, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Soininen, H.; Hiltunen, M. AMP-activated protein kinase: A potential player in Alzheimer’s disease. J. Neurochem. 2011, 118, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Mairet-Coello, G.; Courchet, J.; Pieraut, S.; Courchet, V.; Maximov, A.; Polleux, F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through tau phosphorylation. Neuron 2013, 78, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Che, H.; Sun, L.H.; Guo, F.; Niu, H.F.; Su, X.L.; Bao, Y.N.; Fu, Z.D.; Liu, H.L.; Hou, X.; Yang, B.F.; et al. Expression of amyloid-associated miRNAs in both the forebrain cortex and hippocampus of middle-aged rat. Cell. Physiol. Biochem. 2014, 33, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Y.; Chang, M.N.; Lei, J.X.; Koukiekolo, R.; Smith, B.; Zhang, D.; Ghribi, O. Identification of microRNAs involved in alzheimer’s progression using a rabbit model of the disease. Am. J. Neurodegener. Dis. 2014, 3, 33–44. [Google Scholar] [PubMed]
- Geekiyanage, H.; Jicha, G.A.; Nelson, P.T.; Chan, C. Blood serum miRNA: Non-invasive biomarkers for Alzheimer’s disease. Exp. Neurol. 2012, 235, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yu, J.T.; Liu, Q.Y.; Tan, M.S.; Zhang, W.; Hu, N.; Wang, Y.L.; Sun, L.; Jiang, T. Circulating miR-125b as a biomarker of Alzheimer’s disease. J. Neurol. Sci. 2014, 336, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 253–259. [Google Scholar] [PubMed]
- Krichevsky, A.M.; Sonntag, K.C.; Isacson, O.; Kosik, K.S. Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells 2006, 24, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Kapsimali, M.; Kloosterman, W.P.; de Bruijn, E.; Rosa, F.; Plasterk, R.H.; Wilson, S.W. MicroRNAs show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol. 2007, 8, R173. [Google Scholar] [CrossRef] [PubMed]
- Makeyev, E.V.; Zhang, J.; Carrasco, M.A.; Maniatis, T. The microRNA mir-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 2007, 27, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Al Hashimi, A.; Girard, J.; Delay, C.; Hébert, S.S. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J. Neurochem. 2011, 116, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Estus, S.; Usiak, M.; Younkin, L.H.; Younkin, S.G. Expression of β amyloid protein precursor mRNAs: Recognition of a novel alternatively spliced form and quantitation in Alzheimer’s disease using PCR. Neuron 1990, 4, 253–267. [Google Scholar] [CrossRef]
- Neve, R.L.; Rogers, J.; Higgins, G.A. The alzheimer amyloid precursor-related transcript lacking the β/A4 sequence is specifically increased in Alzheimer’s disease brain. Neuron 1990, 5, 329–338. [Google Scholar] [CrossRef]
- Jacobsen, J.S.; Blume, A.J.; Vitek, M.P. Quantitative measurement of alternatively spliced amyloid precursor protein mRNA expression in Alzheimer’s disease and normal brain by S1 nuclease protection analysis. Neurobiol. Aging 1991, 12, 585–592. [Google Scholar] [CrossRef]
- Fang, M.; Wang, J.; Zhang, X.; Geng, Y.; Hu, Z.; Rudd, J.A.; Ling, S.; Chen, W.; Han, S. The miR-124 regulates the expression of BACE1/β-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 2012, 209, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.; Bossers, K.; Janky, R.; Salta, E.; Frigerio, C.S.; Barbash, S.; Rothman, R.; Sierksma, A.S.; Thathiah, A.; Greenberg, D.; et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 1613–1634. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Maes, O.C.; Chertkow, H.M.; Wang, E. MicroRNA expression in alzheimer blood mononuclear cells. Gene Regul. Syst. Biol. 2007, 1, 263–274. [Google Scholar] [CrossRef]
- Nunez-Iglesias, J.; Liu, C.C.; Morgan, T.E.; Finch, C.E.; Zhou, X.J. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS ONE 2010, 5, e8898. [Google Scholar] [CrossRef] [PubMed]
- Geekiyanage, H.; Chan, C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 2011, 31, 14820–14830. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, E.R.; Kawamoto, E.M.; Taub, D.D.; Lal, A.; Abdelmohsen, K.; Zhang, Y.; Wood, W.H.; Lehrmann, E.; Camandola, S.; Becker, K.G.; et al. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia 2013, 61, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, L.; Gräfe, A.; Seiler, A.; Schumacher, S.; Nitsch, R.; Wulczyn, F.G. Regulation of miRNA expression during neural cell specification. Eur. J. Neurosci. 2005, 21, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, P.; Zhu, H.; Xu, Y.; Ma, C.; Dai, X.; Huang, L.; Liu, Y.; Zhang, L.; Qin, C. MiR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res. Bull. 2009, 80, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Bettens, K.; Brouwers, N.; Engelborghs, S.; Van Miegroet, H.; De Deyn, P.P.; Theuns, J.; Sleegers, K.; Van Broeckhoven, C. App and BACE1 miRNA genetic variability has no major role in risk for Alzheimer disease. Hum. Mutat. 2009, 30, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: MiR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Coy, J.F.; Wiemann, S.; Bechmann, I.; Bächner, D.; Nitsch, R.; Kretz, O.; Christiansen, H.; Poustka, A. Pore membrane and/or filament interacting like protein 1 (POMFIL1) is predominantly expressed in the nervous system and encodes different protein isoforms. Gene 2002, 290, 73–94. [Google Scholar] [CrossRef]
- Hekimi, S.; Kershaw, D. Axonal guidance defects in a Caenorhabditis elegans mutant reveal cell-extrinsic determinants of neuronal morphology. J. Neurosci. 1993, 13, 4254–4271. [Google Scholar] [PubMed]
- Muley, P.D.; McNeill, E.M.; Marzinke, M.A.; Knobel, K.M.; Barr, M.M.; Clagett-Dame, M. The atra-responsive gene neuron navigator 2 functions in neurite outgrowth and axonal elongation. Dev. Neurobiol. 2008, 68, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Kole, A.J.; Swahari, V.; Hammond, S.M.; Deshmukh, M. MiR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev. 2011, 25, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar] [PubMed]
- Giam, M.; Huang, D.C.; Bouillet, P. BH3-only proteins and their roles in programmed cell death. Oncogene 2008, 27 (Suppl. 1), S128–S136. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, K.; Benito, A.; Kiryu-Seo, S.; Gonzalez, V.; Inohara, N.; Lieberman, A.P.; Kiyama, H.; Nuñez, G.; Leiberman, A.P. Critical role for dp5/harakiri, a bcl-2 homology domain 3-only bcl-2 family member, in axotomy-induced neuronal cell death. J. Neurosci. 2004, 24, 3721–3725. [Google Scholar] [CrossRef] [PubMed]
- Putcha, G.V.; Deshmukh, M.; Johnson, E.M. Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J. Cell Biol. 2000, 149, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Wang, H.; Dong, W.; Quan, X.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Qin, C. MiR-29c regulates BACE1 protein expression. Brain Res. 2011, 1395, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Huang, Y.; Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Liyanage, U.; Bickel, P.E.; Xia, W.; Lansbury, P.T.; Kosik, K.S. A detergent-insoluble membrane compartment contains Aβ in vivo. Nat. Med. 1998, 4, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Cheng, H.; Kim, S.H.; Chen, Y.; Barnes, N.Y.; Parent, A.T.; Sisodia, S.S.; Thinakaran, G. Spatial segregation of γ-secretase and substrates in distinct membrane domains. J. Biol. Chem. 2005, 280, 25892–25900. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; Grimm, C.H.; Paya-Cano, J.L.; Sugden, K.; Nietfeld, W.; Lehrach, H.; Schalkwyk, L.C. Using hippocampal microRNA expression differences between mouse inbred strains to characterise miRNA function. Mamm. Genome 2008, 19, 552–560. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.C. Natural cellular inhibitors of caspases. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 215–229. [Google Scholar] [CrossRef]
- Rohn, T.T.; Vyas, V.; Hernandez-Estrada, T.; Nichol, K.E.; Christie, L.A.; Head, E. Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein bcl-2. J. Neurosci. 2008, 28, 3051–3059. [Google Scholar] [CrossRef] [PubMed]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. MicroRNA-34c is a novel target to treat dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. MiR-34a repression of sirt1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Landreh, M.; Cao, K.; Abe, M.; Hendriks, G.J.; Kennerdell, J.R.; Zhu, Y.; Wang, L.S.; Bonini, N.M. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 2012, 482, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, D.; He, Y.; Meléndez, A.; Feng, Z.; Hong, Q.; Bai, X.; Li, Q.; Cai, G.; Wang, J.; et al. MiR-34 modulates Caenorhabditis elegans lifespan via repressing the autophagy gene ATG9. Age 2013, 35, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Dickson, J.R.; Kruse, C.; Montagna, D.R.; Finsen, B.; Wolfe, M.S. Alternative polyadenylation and miR-34 family members regulate Tau expression. J. Neurochem. 2013, 127, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Li, H.; Jiang, L.; Hermeking, H. The p53/miR-34 axis in development and disease. J. Mol. Cell Biol. 2014, 6, 214–230. [Google Scholar] [CrossRef] [PubMed]
- Ohyagi, Y.; Asahara, H.; Chui, D.H.; Tsuruta, Y.; Sakae, N.; Miyoshi, K.; Yamada, T.; Kikuchi, H.; Taniwaki, T.; Murai, H.; et al. Intracellular Aβ42 activates p53 promoter: A pathway to neurodegeneration in alzheimer’s disease. FASEB J. 2005, 19, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of β-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Wang, W.X. Mir-107 is reduced in Alzheimer’s disease brain neocortex: Validation study. J. Alzheimers Dis. 2010, 21, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Van Hoecke, A.; Lambrechts, D.; Vanacker, P.; Bogaert, E.; van Swieten, J.; Carmeliet, P.; Van Den Bosch, L.; Robberecht, W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 2008, 181, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kämäläinen, A.; Viswanathan, J.; Natunen, T.; Helisalmi, S.; Kauppinen, T.; Pikkarainen, M.; Pursiheimo, J.P.; Alafuzoff, I.; Kivipelto, M.; Haapasalo, A.; et al. GRN variant rs5848 reduces plasma and brain levels of granulin in alzheimer’s disease patients. J. Alzheimers Dis. 2013, 33, 23–27. [Google Scholar] [PubMed]
- Wang, W.X.; Wilfred, B.R.; Madathil, S.K.; Tang, G.; Hu, Y.; Dimayuga, J.; Stromberg, A.J.; Huang, Q.; Saatman, K.E.; Nelson, P.T. MiR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am. J. Pathol. 2010, 177, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Muniappan, L.; Tang, G.; Ozcan, S. Identification of glucose-regulated miRNAs from pancreatic β cells reveals a role for miR-30d in insulin transcription. RNA 2009, 15, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Maciver, S.K.; Harrington, C.R. Two actin binding proteins, actin depolymerizing factor and cofilin, are associated with hirano bodies. Neuroreport 1995, 6, 1985–1988. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Hennessey, T.; Flynt, A.; Lai, E.; Beal, M.F.; Lin, M.T. MicroRNA-related cofilin abnormality in Alzheimer’s disease. PLoS ONE 2010, 5, e15546. [Google Scholar] [CrossRef] [PubMed]
- Moncini, S.; Salvi, A.; Zuccotti, P.; Viero, G.; Quattrone, A.; Barlati, S.; De Petro, G.; Venturin, M.; Riva, P. The role of miR-103 and miR-107 in regulation of CDK5R1 expression and in cellular migration. PLoS ONE 2011, 6, e20038. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.F.; Seymour, P.A.; Sanner, M.A.; Schachter, J.B. CDK5 as a drug target for the treatment of Alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 267–273. [Google Scholar] [CrossRef]
- Augustin, R.; Endres, K.; Reinhardt, S.; Kuhn, P.H.; Lichtenthaler, S.F.; Hansen, J.; Wurst, W.; Trümbach, D. Computational identification and experimental validation of microRNAs binding to the Alzheimer-related gene ADAM10. BMC Med. Genet. 2012, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Kuiperij, H.B.; Claassen, J.A.; Küsters, B.; Verbeek, M.M. MicroRNAs in Alzheimer’s disease: Differential expression in hippocampus and cell-free cerebrospinal fluid. Neurobiol. Aging 2014, 35, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Dolios, G.; Wang, R.; Liao, F.F. Soluble β-amyloid peptides, but not insoluble fibrils, have specific effect on neuronal microRNA expression. PLoS ONE 2014, 9, e90770. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Zhao, Y.; Cui, J.G. An Nf-κB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef] [PubMed]
- Granic, I.; Dolga, A.M.; Nijholt, I.M.; van Dijk, G.; Eisel, U.L. Inflammation and NF-κB in Alzheimer’s disease and diabetes. J. Alzheimers Dis. 2009, 16, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Cui, J.G.; Dua, P.; Pogue, A.I.; Bhattacharjee, S.; Lukiw, W.J. Differential expression of miRNA-146a-regulated inflammatory genes in human primary neural, astroglial and microglial cells. Neurosci. Lett. 2011, 499, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Sharma, C.; Hemler, M.E. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 2009, 23, 3674–3681. [Google Scholar] [CrossRef] [PubMed]
- Pogue, A.I.; Percy, M.E.; Cui, J.G.; Li, Y.Y.; Bhattacharjee, S.; Hill, J.M.; Kruck, T.P.; Zhao, Y.; Lukiw, W.J. Up-regulation of Nf-κB-sensitive miRNA-125b and miRNA-146a in metal sulfate-stressed human astroglial (HAG) primary cell cultures. J. Inorg. Biochem. 2011, 105, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. NF-κB-regulated micro RNAs (miRNAs) in primary human brain cells. Exp. Neurol. 2012, 235, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Vilardo, E.; Barbato, C.; Ciotti, M.; Cogoni, C.; Ruberti, F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J. Biol. Chem. 2010, 285, 18344–18351. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Lahiri, D.K. MicroRNA-101 downregulates Alzheimer’s amyloid-β precursor protein levels in human cell cultures and is differentially expressed. Biochem. Biophys. Res. Commun. 2011, 404, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of α-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- Doxakis, E. Post-transcriptional regulation of α-synuclein expression by miR-7 and miR-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-153 physiologically inhibits expression of amyloid-β precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 2012, 287, 31298–31310. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.F.; Sakamoto, K.; Aten, S.; Snider, K.H.; Loeser, J.; Hesse, A.M.; Page, C.E.; Pelz, C.; Arthur, J.S.; Impey, S.; et al. Targeted deletion of miR-132/-212 impairs memory and alters the hippocampal transcriptome. Learn. Mem. 2016, 23, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E.; et al. MiR-132/212 deficiency impairs Tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.B.; Mufson, E.J.; Counts, S.E. Evidence for a neuroprotective microRNA pathway in amnestic mild cognitive impairment. Front. Neurosci. 2015, 9, 430. [Google Scholar] [CrossRef] [PubMed]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Bobbin, M.L.; Rossi, J.J. RNA interference (RNAi)-based therapeutics: Delivering on the promise? Annu. Rev. Pharmacol. Toxicol. 2016, 56, 103–122. [Google Scholar] [CrossRef] [PubMed]
- Mack, G.S. MicroRNA gets down to business. Nat. Biotechnol. 2007, 25, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Landthaler, M.; Dorsett, Y.; Tuschl, T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA 2004, 10, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.E.; Nuovo, G.J.; Terry, A.V., Jr.; Martin, M.M.; Malana, G.E.; Sansom, S.E.; Pleister, A.P.; Beck, W.D.; Head, E.; Feldman, D.S.; et al. Chromosome 21-derived microRNAs provide an etiological basis for aberrant protein expression in human down syndrome brains. J. Biol. Chem. 2010, 285, 1529–1543. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Kauppinen, S.; Lund, A.H. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene 2006, 372, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Chugh, P.; Dittmer, D.P. Potential pitfalls in microRNA profiling. Wiley Interdiscip. Rev. RNA 2012, 3, 601–616. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| microRNA | Evidence of Up-Regulation in AD Brain | Evidence of Down-Regulation in AD Brain | AD-Relevant Targets | AD-Relevant Regulators |
|---|---|---|---|---|
| miR-9 | ||||
| miR-124 | N/A |
| ||
| miR-181 | N/A |
| ||
| miR-29 |
| N/A | ||
| miR-34 | N/A | N/A | ||
| miR-107 | N/A |
| ||
| miR-146 |
| N/A | ||
| miR-101 | N/A |
| N/A | |
| miR-195 |
|
|
| N/A |
| miR-153 | N/A |
| N/A |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miya Shaik, M.; Tamargo, I.A.; Abubakar, M.B.; Kamal, M.A.; Greig, N.H.; Gan, S.H. The Role of microRNAs in Alzheimer’s Disease and Their Therapeutic Potentials. Genes 2018, 9, 174. https://doi.org/10.3390/genes9040174
Miya Shaik M, Tamargo IA, Abubakar MB, Kamal MA, Greig NH, Gan SH. The Role of microRNAs in Alzheimer’s Disease and Their Therapeutic Potentials. Genes. 2018; 9(4):174. https://doi.org/10.3390/genes9040174
Chicago/Turabian StyleMiya Shaik, Munvar, Ian A. Tamargo, Murtala B. Abubakar, Mohammad A. Kamal, Nigel H. Greig, and Siew Hua Gan. 2018. "The Role of microRNAs in Alzheimer’s Disease and Their Therapeutic Potentials" Genes 9, no. 4: 174. https://doi.org/10.3390/genes9040174
APA StyleMiya Shaik, M., Tamargo, I. A., Abubakar, M. B., Kamal, M. A., Greig, N. H., & Gan, S. H. (2018). The Role of microRNAs in Alzheimer’s Disease and Their Therapeutic Potentials. Genes, 9(4), 174. https://doi.org/10.3390/genes9040174