The Genetic Architecture Underlying the Evolution of a Rare Piscivorous Life History Form in Brown Trout after Secondary Contact and Strong Introgression

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Library Preparation and Sequencing
2.3. ddRAD Genotyping and Bioinformatics
2.4. Population Genomic Analyses
2.5. Correlation between Ancestry Coefficient and Morphology
2.6. Inferring the Divergence History of Benthivorous Brown Trout and Ferox Trout Evolution
2.7. Genome-Scans and Genomic Outlier Regions
2.8. Outlier Annotation and Over-Representation Analysis
2.9. Mitochondrial DNA: Sequencing and Analysis of the ND1 Gene
3. Results
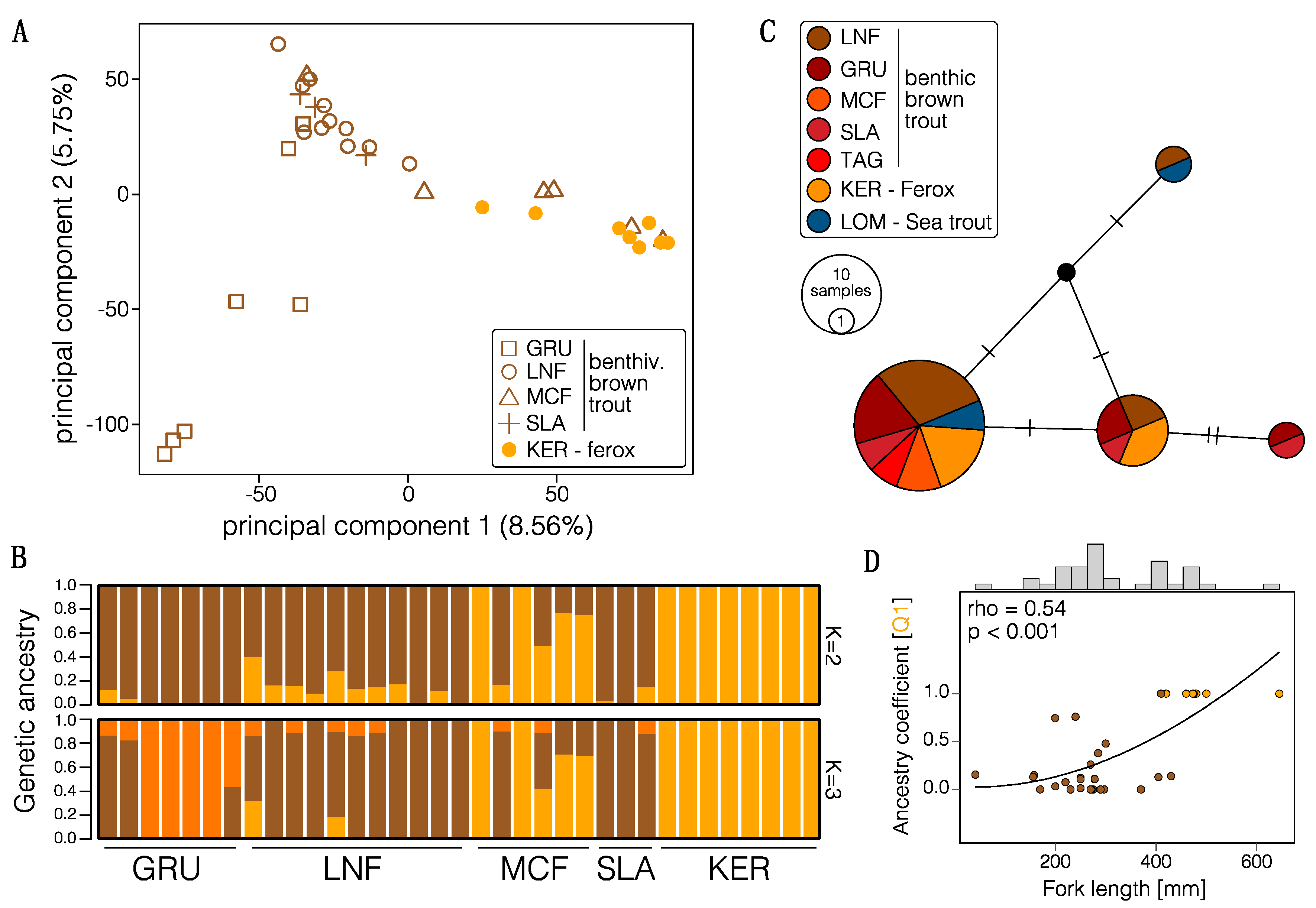
3.1. Genome-Wide Dataset and Summary Statistics
3.2. Population Structure and Admixture
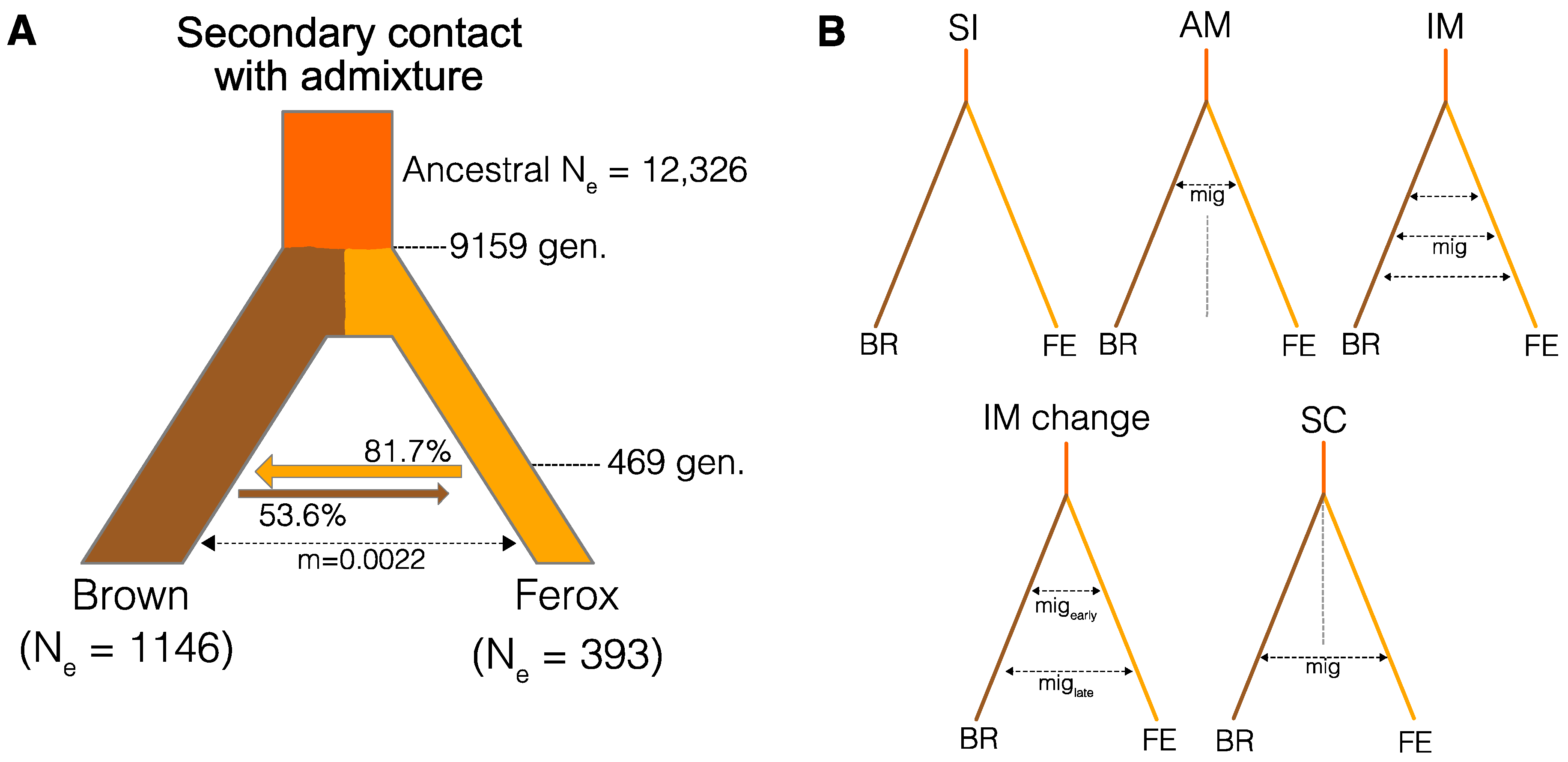
3.3. Divergence History of Benthivorous Brown Trout and Ferox Trout
3.4. Outlier Analysis and Genome-Scans
3.5. Annotation of Pcadapt Outlier Loci and Genomic Outlier Windows
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Turner, T.L.; Hahn, M.W.; Nuzhdin, S.V. Genomic Islands of Speciation in Anopheles gambiae. PLoS Biol. 2005, 3, e285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brawand, D.; Wagner, C.E.; Li, Y.I.; Malinsky, M.; Keller, I.; Fan, S.; Simakov, O.; Ng, A.Y.; Lim, Z.W.; Bezault, E.; et al. The genomic substrate for adaptive radiation in African cichlid fish. Nature 2014, 513, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinsky, M.; Challis, R.J.; Tyers, A.M.; Schiffels, S.; Terai, Y.; Ngatunga, B.P.; Miska, E.A.; Durbin, R.; Genner, M.J.; Turner, G.F. Genomic islands of speciation separate cichlid ecomorphs in an East African crater lake. Science 2015, 350, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Harr, B. Genomic islands of differentiation between house mouse subspecies. Genome Res. 2006, 16, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, N.J.; Whibley, A.; Jones, R.T.; Davey, J.W.; Dasmahapatra, K.K.; Baxter, S.W.; Quail, M.A.; Joron, M.; Ffrench-Constant, R.H.; Blaxter, M.L.; et al. Genomic islands of divergence in hybridizing Heliconius butterflies identified by large-scale targeted sequencing. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Renaut, S.; Maillet, N.; Normandeau, E.; Sauvage, C.; Derome, N.; Rogers, S.M.; Bernatchez, L. Genome-wide patterns of divergence during speciation: The lake whitefish case study. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2012, 367, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Gagnaire, P.-A.; Pavey, S.A.; Normandeau, E.; Bernatchez, L. The genetic architecture of reproductive isolation during speciation-with-gene-flow in lake whitefish species pairs assessed by RAD sequencing. Evolution 2013, 67, 2483–2497. [Google Scholar] [CrossRef] [PubMed]
- Soria-Carrasco, V.; Gompert, Z.; Comeault, A.A.; Farkas, T.E.; Parchman, T.L.; Johnston, J.S.; Buerkle, C.A.; Feder, J.L.; Bast, J.; Schwander, T.; et al. Stick insect genomes reveal natural selection’s role in parallel speciation. Science 2014, 344, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Marques, D.A.; Lucek, K.; Meier, J.I.; Mwaiko, S.; Wagner, C.E.; Excoffier, L.; Seehausen, O. Genomics of Rapid Incipient Speciation in Sympatric Threespine Stickleback. PLoS Genet. 2016, 12, e1005887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riesch, R.; Muschick, M.; Lindtke, D.; Villoutreix, R.; Comeault, A.A.; Farkas, T.E.; Lucek, K.; Hellen, E.; Soria-Carrasco, V.; Dennis, S.R.; et al. Transitions between phases of genomic differentiation during stick-insect speciation. Nat. Ecol. Evol. 2017, 1, 82. [Google Scholar] [CrossRef] [PubMed]
- Larson, W.A.; Limborg, M.T.; McKinney, G.J.; Schindler, D.E.; Seeb, J.E.; Seeb, L.W. Genomic islands of divergence linked to ecotypic variation in sockeye salmon. Mol. Ecol. 2017, 26, 554–570. [Google Scholar] [CrossRef] [PubMed]
- Seehausen, O.; Butlin, R.K.; Keller, I.; Wagner, C.E.; Boughman, J.W.; Hohenlohe, P.A.; Peichel, C.L.; Saetre, G.-P.; Bank, C.; Brännström, A.; et al. Genomics and the origin of species. Nat. Rev. Genet. 2014, 15, 176–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, J.B.W.; Ellegren, H. Making sense of genomic islands of differentiation in light of speciation. Nat. Rev. Genet. 2016, 18, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Ravinet, M.; Faria, R.; Butlin, R.K.; Galindo, J.; Bierne, N.; Rafajlović, M.; Noor, M.A.F.; Mehlig, B.; Westram, A.M. Interpreting the genomic landscape of speciation: A road map for finding barriers to gene flow. J. Evol. Biol. 2017, 30, 1450–1477. [Google Scholar] [CrossRef] [PubMed]
- Burri, R. Interpreting differentiation landscapes in the light of long-term linked selection. Evol. Lett. 2017, 1, 118–131. [Google Scholar] [CrossRef]
- Berner, D.; Roesti, M. Genomics of adaptive divergence with chromosome-scale heterogeneity in crossover rate. Mol. Ecol. 2017, 26, 6351–6369. [Google Scholar] [CrossRef] [PubMed]
- Noor, M.A.F.; Bennett, S.M. Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species. Heredity 2009, 103, 439. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, T.E.; Hahn, M.W. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 2014, 23, 3133–3157. [Google Scholar] [CrossRef] [PubMed]
- Renaut, S.; Owens, G.L.; Rieseberg, L.H. Shared selective pressure and local genomic landscape lead to repeatable patterns of genomic divergence in sunflowers. Mol. Ecol. 2014, 23, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Burri, R.; Nater, A.; Kawakami, T.; Mugal, C.F.; Olason, P.I.; Smeds, L.; Suh, A.; Dutoit, L.; Bure, S.; Garamszegi, L.Z.; et al. Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res. 2015, 25, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.F.; Hahn, M.W. Speciation as a sieve for ancestral polymorphism. Mol. Ecol. 2017, 26, 5362–5368. [Google Scholar] [CrossRef] [PubMed]
- Bay, R.A.; Ruegg, K. Genomic islands of divergence or opportunities for introgression? Proc. Biol. Sci. 2017, 284. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.L.; Hahn, M.W. Genomic islands of speciation or genomic islands and speciation? Mol. Ecol. 2010, 19, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Street, N.R.; Scofield, D.G.; Ingvarsson, P.K. Variation in linked selection and recombination drive genomic divergence during allopatric speciation of European and American aspens. Mol. Biol. Evol. 2016, 33, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Lamichhaney, S.; Grant, R.B.; Grant, P.R.; Andersson, L.; Webster, M.T. Gene flow, ancient polymorphism, and ecological adaptation shape the genomic landscape of divergence among Darwin’s finches. Genome Res. 2017, 27, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.L.; Flaxman, S.M.; Egan, S.P.; Comeault, A.A.; Nosil, P. Geographic Mode of Speciation and Genomic Divergence. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 73–97. [Google Scholar] [CrossRef]
- Yeaman, S.; Aeschbacher, S.; Bürger, R. The evolution of genomic islands by increased establishment probability of linked alleles. Mol. Ecol. 2016, 25, 2542–2558. [Google Scholar] [CrossRef] [PubMed]
- Sicard, A.; Kappel, C.; Josephs, E.B.; Lee, Y.; Marona, C.; Stinchcombe, J.R.; Wright, S.I.; Lenhard, M. Divergent sorting of a balanced ancestral polymorphism underlies the establishment of gene-flow barriers in Capsella. Nat. Commun. 2015, 6, 7960. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Wang, K.; Hu, Q.; Xi, Z.; Wan, D.; Wang, Q.; Feng, J.; Jiang, D.; Ahani, H.; Abbott, R.J.; et al. Ancient polymorphisms and divergence hitchhiking contribute to genomic islands of divergence within a poplar species complex. Proc. Natl. Acad. Sci. USA 2017, 115, E236–E243. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, B.; Jonsson, N. Polymorphism and speciation in Arctic charr. J. Fish Biol. 2001, 58, 605–638. [Google Scholar] [CrossRef]
- Wood, C.C.; Bickham, J.W.; John Nelson, R.; Foote, C.J.; Patton, J.C. Recurrent evolution of life history ecotypes in sockeye salmon: Implications for conservation and future evolution. Evol. Appl. 2008, 1, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, L.; Renaut, S.; Whiteley, A.R.; Derome, N.; Jeukens, J.; Landry, L.; Lu, G.; Nolte, A.W.; Ostbye, K.; Rogers, S.M.; et al. On the origin of species: Insights from the ecological genomics of lake whitefish. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2010, 365, 1783–1800. [Google Scholar] [CrossRef] [PubMed]
- Præbel, K.; Knudsen, R.; Siwertsson, A.; Karhunen, M.; Kahilainen, K.K.; Ovaskainen, O.; Østbye, K.; Peruzzi, S.; Fevolden, S.E.; Amundsen, P.A. Ecological speciation in postglacial European whitefish: Rapid adaptive radiations into the littoral, pelagic, and profundal lake habitats. Ecol. Evol. 2013, 3, 4970–4986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, A.M.; Hansen, M.J.; Bronte, C.R.; Krueger, C.C. If Arctic charr Salvelinus alpinus is ‘the most diverse vertebrate’, what is the lake charr Salvelinus namaycush? Fish Fish. 2015, 17, 1194–1207. [Google Scholar] [CrossRef]
- Gordeeva, N.V.; Alekseyev, S.S.; Matveev, A.N.; Samusenok, V.P. Parallel evolutionary divergence in Arctic charr Salvelinus alpinus (L.) complex from Transbaikalia: Variation in differentiation degree and segregation of genetic diversity between sympatric forms. Can. J. Fish. Aquat. Sci. 2015, 72, 96–115. [Google Scholar] [CrossRef]
- Hudson, A.G.; Vonlanthen, P.; Seehausen, O. Rapid parallel adaptive radiations from a single hybridogenic ancestral population. Proc. R. Soc. B Biol. Sci. 2011, 278, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Rougeux, C.; Bernatchez, L.; Gagnaire, P.-A.A. Modeling the Multiple Facets of Speciation-with-Gene-Flow toward Inferring the Divergence History of Lake Whitefish Species Pairs (Coregonus clupeaformis). Genome Biol. Evol. 2017, 9, 2057–2074. [Google Scholar] [CrossRef] [PubMed]
- Klemetsen, A.; Amundsen, P.-A.; Dempson, J.B.; Jonsson, B.; Jonsson, N.; O’Connell, M.F.; Mortensen, E. Atlantic salmon Salmo salar L., brown trout Salmo trutta L. and Arctic charr Salvelinus alpinus (L.): A review of aspects of their life histories. Ecol. Freshw. Fish 2003, 12, 1–59. [Google Scholar] [CrossRef]
- Campbell, R.N. Ferox trout, Salmo trutta L., and charr, Salvelinus alpinus (L.), in Scottish lochs. J. Fish Biol. 1979, 14, 1–29. [Google Scholar] [CrossRef]
- Ferguson, A.; Mason, F.M. Allozyme evidence for reproductively isolated sympatric populations of brown trout Salmo trutta L. in Lough Melvin, Ireland. J. Fish Biol. 1981, 18, 629–642. [Google Scholar] [CrossRef]
- Greer, R. Ferox Trout and Arctic Charr: A Predator, Its Pursuit and Its Prey; Swan-Hill Press: Shrewsbury, UK, 1995. [Google Scholar]
- Hughes, M.R.; Dodd, J.A.; Maitland, P.S.; Adams, C.E. Lake bathymetry and species occurrence predict the distribution of a lacustrine apex predator. J. Fish Biol. 2016, 88, 1648–1654. [Google Scholar] [CrossRef] [PubMed]
- Alekseyev, S.S.; Samusenok, V.P.; Matveev, A.N.; Yu, M. Diversification, sympatric speciation, and trophic polymorphism of Arctic charr, Salvelinus alpinus complex, in Transbaikalia. Environ. Biol. Fishes 2002, 64, 97–114. [Google Scholar] [CrossRef]
- Zimmerman, M.S.; Schmidt, S.N.; Krueger, C.C.; Vander Zanden, J.M.; Eshenroder, R.L. Ontogenetic niche shifts and resource partitioning of lake trout morphotypes. Can. J. Fish. Aquat. Sci. 2009, 66, 1007–1018. [Google Scholar] [CrossRef]
- Smalås, A.; Amundsen, P.-A.; Knudsen, R. Contrasting life history strategies of sympatric Arctic charr morphs, Salvelinus alpinus. J. Ichthyol. 2013, 53, 856–866. [Google Scholar] [CrossRef]
- Thorne, A.; MacDonald, A.I.; Thorley, J.L. The abundance of large, piscivorous Ferox Trout (Salmo trutta) in Loch Rannoch, Scotland. PeerJ 2016, 4, e2646. [Google Scholar] [CrossRef] [PubMed]
- Grey, J. Ontogeny and dietary specialization in brown trout (Salmo trutta L.) from Loch Ness, Scotland, examined using stable isotopes of carbon and nitrogen. Ecol. Freshw. Fish 2001, 10, 168–176. [Google Scholar] [CrossRef]
- Grey, J.; Thackeray, S.J.; Jones, R.I.; Shine, A. Ferox Trout (Salmo trutta) as ‘Russian dolls’: Complementary gut content and stable isotope analyses of the Loch Ness foodweb. Freshw. Biol. 2002, 47, 1235–1243. [Google Scholar] [CrossRef]
- Hughes, M.R.; Van Leeuwen, T.E.; Cunningham, P.D.; Adams, C.E. Parentally acquired differences in resource acquisition ability between brown trout from alternative life history parentage. Ecol. Freshw. Fish 2016, 27, 62–69. [Google Scholar] [CrossRef]
- Mangel, M. Life history invariants, age at maturity and the ferox trout. Evol. Ecol. 1996, 10, 249–263. [Google Scholar] [CrossRef]
- Mangel, M.; Abrahams, M.V. Age and longevity in fish, with consideration of the ferox trout. Exp. Gerontol. 2001, 36, 765–790. [Google Scholar] [CrossRef]
- Campbell, R.N. The growth of brown trout Salmo trutta L. in northern Scottish lochs with special reference to the improvement of fisheries. J. Fish Biol. 1971, 3, 1–28. [Google Scholar] [CrossRef]
- Persson, L.; Andersson, J.; Wahlstrom, E.; Eklov, P. Size-Specific Interactions in Lake Systems: Predator Gape Limitation and prey Growth Rate and Mortality. Ecology 1996, 77, 900–911. [Google Scholar] [CrossRef]
- Mittelbach, G.G.; Persson, L. The ontogeny of piscivory and its ecological consequences. Can. J. Fish. Aquat. Sci. 1998, 55, 1454–1465. [Google Scholar] [CrossRef]
- Ferguson, A.; Taggart, J.B. Genetic differentiation among the sympatric brown trout (Salmo trutta) populations of Lough Melvin, Ireland. Biol. J. Linn. Soc. 1991, 43, 221–237. [Google Scholar] [CrossRef]
- Prodöhl, P.A.; Taggart, J.B.; Ferguson, A. Genetic variability within and among sympatric brown trout (Salmo trutta) populations: Multi-locus DNA fingerprint analysis. Hereditas 1992, 117, 45–50. [Google Scholar] [CrossRef]
- McVeigh, H.P.; Hynes, R.A.; Ferguson, A. Mitochondrial DNA differentiation of sympatric populations of brown trout, Salmo trutta L., from Lough Melvin, Ireland. Can. J. Fish. Aquat. Sci. 1995, 52, 1617–1622. [Google Scholar] [CrossRef]
- Duguid, R.A.; Ferguson, A.; Prodöhl, P. Reproductive isolation and genetic differentiation of ferox trout from sympatric brown trout in Loch Awe and Loch Laggan, Scotland. J. Fish Biol. 2006, 69, 89–114. [Google Scholar] [CrossRef] [Green Version]
- McKeown, N.J.; Hynes, R.A.; Duguid, R.A.; Ferguson, A.; Prodöhl, P.A. Phylogeographic structure of brown trout Salmo trutta in Britain and Ireland: Glacial refugia, postglacial colonization and origins of sympatric populations. J. Fish Biol. 2010, 76, 319–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freyhof, J.; Kottelat, M. Salmo ferox. The IUCN Red List of Threatened Species 2008: e.T135577A41506832008; International Union for Conservation of Nature and Natural Resources: Gland, Switzerland, 2008. [Google Scholar]
- Orr, H.A.; Turelli, M. The evolution of postzygotic isolation: Accumulating Dobzhansky-Muller incompatibilities. Evolution 2001, 55, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.M.; Bernatchez, L. The genetic basis of intrinsic and extrinsic post-zygotic reproductive isolation jointly promoting speciation in the lake whitefish species complex (Coregonus clupeaformis). J. Evol. Biol. 2006, 19, 1979–1994. [Google Scholar] [CrossRef] [PubMed]
- Schluter, D. Evidence for Ecological Speciation and Its Alternative. Science 2009, 323, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Seehausen, O.; Wagner, C.E. Speciation in Freshwater Fishes. Annu. Rev. Ecol. Evol. Syst. 2014, 45, 621–651. [Google Scholar] [CrossRef]
- Lu, G.; Bernatchez, L. Experimental evidence for reduced hybrid viability between dwarf and normal ecotypes of lake whitefish (Coregonus clupeaformis Mitchill). Proc. R. Soc. B Biol. Sci. 1998, 265, 1025–1030. [Google Scholar] [CrossRef]
- Eek, D.; Bohlin, T. Strontium in scales verifies that sympatric sea-run and stream-resident brown trout can be distinguished by coloration. J. Fish Biol. 1997, 51, 659–661. [Google Scholar] [CrossRef]
- Recknagel, H.; Jacobs, A.; Herzyk, P.; Elmer, K.R. Double-digest RAD sequencing using Ion Proton semiconductor platform (ddRADseq-ion) with nonmodel organisms. Mol. Ecol. Resour. 2015, 15, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H.; Koning, D.-J. De Stacks: Building and Genotyping Loci De Novo from Short-Read Sequences. G3 Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Ben, F.K.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Macqueen, D.J.; Primmer, C.R.; Houston, R.D.; Nowak, B.F.; Bernatchez, L.; Bergseth, S.; Davidson, W.S.; Gallardo-Escárate, C.; Goldammer, T.; Guiguen, Y.; et al. Functional Annotation of All Salmonid Genomes (FAASG): An international initiative supporting future salmonid research, conservation and aquaculture. BMC Genom. 2017, 18, 484. [Google Scholar] [CrossRef] [PubMed]
- Leitwein, M.; Guinand, B.; Pouzadoux, J.; Desmarais, E.; Berrebi, P.; Gagnaire, P.-A.A. Dense Brown Trout (Salmo trutta) Linkage Map Reveals Recent Chromosomal Rearrangements in the Salmo Genus and the Impact of Selection on Linked Neutral Diversity. G3 Genes Genomes Genet. 2017, 7, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.B.; Keatley, K.A.; Morasch, M.R.; Ventura, A.B.; Lubieniecki, K.P.; Koop, B.F.; Danzmann, R.G.; Davidson, W.S. Assignment of Atlantic salmon (Salmo salar) linkage groups to specific chromosomes: Conservation of large syntenic blocks corresponding to whole chromosome arms in rainbow trout (Oncorhynchus mykiss). BMC Genet. 2009, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Guyomard, R.; Boussaha, M.; Krieg, F.; Hervet, C.; Quillet, E. A synthetic rainbow trout linkage map provides new insights into the salmonid whole genome duplication and the conservation of synteny among teleosts. BMC Genet. 2012, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Leitwein, M.; Gagnaire, P.-A.; Desmarais, E.; Guendouz, S.; Rohmer, M.; Berrebi, P.; Guinand, B. Genome-wide nucleotide diversity of hatchery-reared Atlantic and Mediterranean strains of brown trout Salmo trutta compared to wild Mediterranean populations. J. Fish Biol. 2016, 89, 2717–2734. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G.; Tienderen, P.H. genotype and genodive: Two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 2004, 4, 792–794. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Estimating phylogenetic trees and networks using SplitsTree 4. 2005; manuscript in preparation. [Google Scholar]
- Excoffier, L.; Dupanloup, I.; Huerta-Sánchez, E.; Sousa, V.C.; Foll, M. Robust Demographic Inference from Genomic and SNP Data. PLoS Genet. 2013, 9, e1003905. [Google Scholar] [CrossRef] [PubMed]
- Gutenkunst, R.N.; Hernandez, R.D.; Williamson, S.H.; Bustamante, C.D. Inferring the Joint Demographic History of Multiple Populations from Multidimensional SNP Frequency Data. PLoS Genet. 2009, 5, e1000695. [Google Scholar] [CrossRef] [PubMed]
- Kautt, A.F.; Machado-Schiaffino, G.; Meyer, A. Multispecies Outcomes of Sympatric Speciation after Admixture with the Source Population in Two Radiations of Nicaraguan Crater Lake Cichlids. PLoS Genet. 2016, 12, e1006157. [Google Scholar] [CrossRef] [PubMed]
- Tine, M.; Kuhl, H.; Gagnaire, P.-A.; Louro, B.; Desmarais, E.; Martins, R.S.T.; Hecht, J.; Knaust, F.; Belkhir, K.; Klages, S.; et al. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat. Commun. 2014, 5, 5770. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.I.; Sousa, V.C.; Marques, D.A.; Selz, O.M.; Wagner, C.E.; Excoffier, L.; Seehausen, O. Demographic modelling with whole-genome data reveals parallel origin of similar Pundamilia cichlid species after hybridization. Mol. Ecol. 2017, 26, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Egger, B.; Rösti, M.; Böhne, A.; Roth, O.; Salzburger, W. Demography and genome divergence of lake and stream populations of an East African cichlid fish. Mol. Ecol. 2017, 26, 5016–5030. [Google Scholar] [CrossRef] [PubMed]
- Hesterberg, T. Resample: Resampling Functions. R Package version 0.4. 2015. Available online: https//CRAN.R-project.org/web/packages/resample (accessed on 24 November 2017).
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Bassham, S.; Catchen, J.; Lescak, E.; von Hippel, F.A.; Cresko, W.A. Sweeping genomic remodeling through repeated selection of alternatively adapted haplotypes occurs in the first decades after marine stickleback colonize new freshwater ponds. bioRxiv 2017, 191627. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Catchen, J.; Cresko, W.A. Data Production and Analysis in Population Genomics. Methods Mol. Biol. 2012, 888, 235–260. [Google Scholar] [CrossRef] [PubMed]
- Luu, K.; Bazin, E.; Blum, M.G. pcadapt: An R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 2017, 17, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Duforet-Frebourg, N.; Bazin, E.; Blum, M.G. Genome scans for detecting footprints of local adaptation using a Bayesian factor model. Mol. Biol. Evol. 2014, 31, 2483–2495. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-B.; Lee, S.H.; Zhu, Z.-X.; Benyamin, B.; Robinson, M.R. EigenGWAS: Finding loci under selection through genome-wide association studies of eigenvectors in structured populations. Heredity 2016, 117, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Fariello, M.; Boitard, S.; Naya, H.; SanCristobal, M.; Servin, B. Detecting Signatures of Selection through Haplotype Differentiation among Hierarchically Structured Populations. Genetics 2013, 193, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.; Womack, R.; Chen, M.; Gharbi, K.; Elmer, K. Significant Synteny and Co-localization of Ecologically Relevant Quantitative Trait Loci within and Across Species of Salmonid Fishes. Genetics 2017, 207, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Schenekar, T.; Lerceteau-Köhler, E.; Weiss, S. Fine-scale phylogeographic contact zone in Austrian brown trout Salmo trutta reveals multiple waves of post-glacial colonization and a pre-dominance of natural versus anthropogenic admixture. Conserv. Genet. 2014, 15, 561–572. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Hendry, A.P.; Wenburg, J.K.; Bentzen, P.; Volk, E.C.; Quinn, T.P. Rapid Evolution of Reproductive Isolation in the Wild: Evidence from Introduced Salmon. Science 2000, 290, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Bernatchez, L. Correlated Trophic Specialization and Genetic Divergence in Sympatric Lake Whitefish Ecotypes (Coregonus clupeaformis): Support for the Ecological Speciation Hypothesis. Evolution 1999, 53, 1491–1505. [Google Scholar] [PubMed]
- Østbye, K.; Amundsen, P.-A.; Bernatchez, L.; Klemetsen, A.; Knudsen, R.; Kristoffersen, R.; Naesje, T.F.; Hindar, K. Parallel evolution of ecomorphological traits in the European whitefish Coregonus lavaretus (L.) species complex during postglacial times. Mol. Ecol. 2006, 15, 3983–4001. [Google Scholar] [CrossRef] [PubMed]
- Schluter, D. Ecological Speciation in Postglacial Fishes. Philos. Trans. R. Soc. B Biol. Sci. 1996, 351, 807–814. [Google Scholar] [CrossRef]
- Serbezov, D.; Bernatchez, L.; Olsen, E.M.; Vøllestad, L.A. Mating patterns and determinants of individual reproductive success in brown trout (Salmo trutta) revealed by parentage analysis of an entire stream living population. Mol. Ecol. 2010, 19, 3193–3205. [Google Scholar] [CrossRef] [PubMed]
- Chiverrell, R.C.; Thomas, G.S.P. Extent and timing of the Last Glacial Maximum (LGM) in Britain and Ireland: A review. J. Quat. Sci. 2010, 25, 535–549. [Google Scholar] [CrossRef]
- Bernatchez, L.; Glémet, H.; Wilson, C.C.; Danzmann, R.G. Introgression and fixation of Arctic char (Salvelinus alpinus) mitochondrial genome in an allopatric population of brook trout (Salvelinus fontinalis). Can. J. Fish. Aquat. Sci. 1995, 52, 179–185. [Google Scholar] [CrossRef]
- Quinn, T.P.; Foote, C.J. The effects of body size and sexual dimorphism on the reproductive behaviour of sockeye salmon, Oncorhynchus nerka. Anim. Behav. 1994, 48, 751–761. [Google Scholar] [CrossRef]
- Bierne, N.; Gagnaire, P.A.; David, P. The geography of introgression in a patchy environment and the thorn in the side of ecological speciation. Curr. Zool. 2013, 59, 72–86. [Google Scholar] [CrossRef]
- Vijay, N.; Bossu, C.M.; Poelstra, J.W.; Weissensteiner, M.H.; Suh, A.; Kryukov, A.P.; Wolf, J.B.W. Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat. Commun. 2016, 7, 13195. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009, 10, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Hindar, K.; Jonsson, B. Ecological polymorphism in Arctic charr. Biol. J. Linn. Soc. 1993, 48, 63–74. [Google Scholar] [CrossRef]
- Weber, J.N.; Steinel, N.C.; Shim, K.C.; Bolnick, D.I. Recent evolution of extreme cestode growth suppression by a vertebrate host. Proc. Natl. Acad. Sci. USA 2017, 114, 6575–6580. [Google Scholar] [CrossRef] [PubMed]
- Hooker, O.E.; Barry, J.; Van Leeuwen, T.E.; Lyle, A.; Newton, J.; Cunningham, P.; Adams, C.E. Morphological, ecological and behavioural differentiation of sympatric profundal and pelagic Arctic charr (Salvelinus alpinus) in Loch Dughaill Scotland. Hydrobiologia 2016, 783, 209–221. [Google Scholar] [CrossRef]
- Barry, J.; Newton, M.; Dodd, J.A.; Evans, D.; Newton, J.; Adams, C.E. The effect of foraging and ontogeny on the prevalence and intensity of the invasive parasite Anguillicola crassus in the European eel Anguilla anguilla. J. Fish Dis. 2017, 40, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Elmer, K.R. Genomic tools for new insights to variation, adaptation, and evolution in the salmonid fishes: A perspective for charr. Hydrobiologia 2016, 783, 191–208. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Code | Life History | N. ddRAD | N. mtDNA | Private | SNPs | HO | π |
|---|---|---|---|---|---|---|---|---|
| Kernsary | KER | ferox | 8 | 8 | 99 | 15,698 | 0.005 | 0.006 |
| McFarlane | MCF | bn | 6 | 6 | 48 | 15,802 | 0.006 | 0.006 |
| Slattadale | SLA | bn | 3 | 3 | 20 | 15,219 | 0.005 | 0.006 |
| Loch na Fideil | LNF | bn | 11 | 11 | 197 | 15,644 | 0.006 | 0.006 |
| Grudie | GRU | bn | 7 | 7 | 111 | 15,488 | 0.006 | 0.006 |
| Taggan | TAG | bn | - | 3 | - | - | - | - |
| Loch Lomond | STL | sea trout | - | 3 | - | - | - | - |
| Marker | Source of Variation | Nested in | % var | F-Stat | F-Value | p-Value |
|---|---|---|---|---|---|---|
| ddRADseq | Within ind. | - | 85.5 | Fit | 0.145 | - |
| Among ind. | Pop | 4.3 | Fis | 0.048 | <0.001 | |
| Among sites 1 | Type | 7.2 | Fsc | 0.074 | <0.001 | |
| Among types 2 | - | 3.0 | Fct | 0.030 | 0.188 |
| Model | ln(lhoods) 1 | N. Parameters | AIC 2 | ∆AIC |
|---|---|---|---|---|
| SCadm | −109,160.64 | 8 | 218,337.28 | 0 |
| IMchange | −109,198.39 | 7 | 218,410.79 | 73.5 |
| SC | −109,221.83 | 6 | 218,455.65 | 118.4 |
| IM | −109,228.29 | 5 | 218,466.57 | 129.3 |
| AM | −109,230.45 | 6 | 218,472.90 | 135.6 |
| SI | −109,679.11 | 4 | 219,366.21 | 1028.9 |
| Parameter | Mean | 2.5% CI | 97.5% CI |
|---|---|---|---|
| Ancestral Ne | 12,326 | 7955 | 16,250 |
| Ne brown | 1146 | 1110 | 1173 |
| Ne ferox | 393 | 376 | 409 |
| adm (F-B) 1 | 0.817 | 0.806 | 0.827 |
| adm (B-F) 2 | 0.536 | 0.418 | 0.648 |
| Mig 3 | 0.00220 | 0.00210 | 0.00232 |
| Tsc 4 | 469 | 445 | 498 |
| Tdiv 5 | 9159 | 8271 | 10,292 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacobs, A.; Hughes, M.R.; Robinson, P.C.; Adams, C.E.; Elmer, K.R. The Genetic Architecture Underlying the Evolution of a Rare Piscivorous Life History Form in Brown Trout after Secondary Contact and Strong Introgression. Genes 2018, 9, 280. https://doi.org/10.3390/genes9060280
Jacobs A, Hughes MR, Robinson PC, Adams CE, Elmer KR. The Genetic Architecture Underlying the Evolution of a Rare Piscivorous Life History Form in Brown Trout after Secondary Contact and Strong Introgression. Genes. 2018; 9(6):280. https://doi.org/10.3390/genes9060280
Chicago/Turabian StyleJacobs, Arne, Martin R. Hughes, Paige C. Robinson, Colin E. Adams, and Kathryn R. Elmer. 2018. "The Genetic Architecture Underlying the Evolution of a Rare Piscivorous Life History Form in Brown Trout after Secondary Contact and Strong Introgression" Genes 9, no. 6: 280. https://doi.org/10.3390/genes9060280
APA StyleJacobs, A., Hughes, M. R., Robinson, P. C., Adams, C. E., & Elmer, K. R. (2018). The Genetic Architecture Underlying the Evolution of a Rare Piscivorous Life History Form in Brown Trout after Secondary Contact and Strong Introgression. Genes, 9(6), 280. https://doi.org/10.3390/genes9060280