Efficient Knock-in of a Point Mutation in Porcine Fibroblasts Using the CRISPR/Cas9-GMNN Fusion Gene


 ,
,
Abstract
:1. Introduction
2. Methods
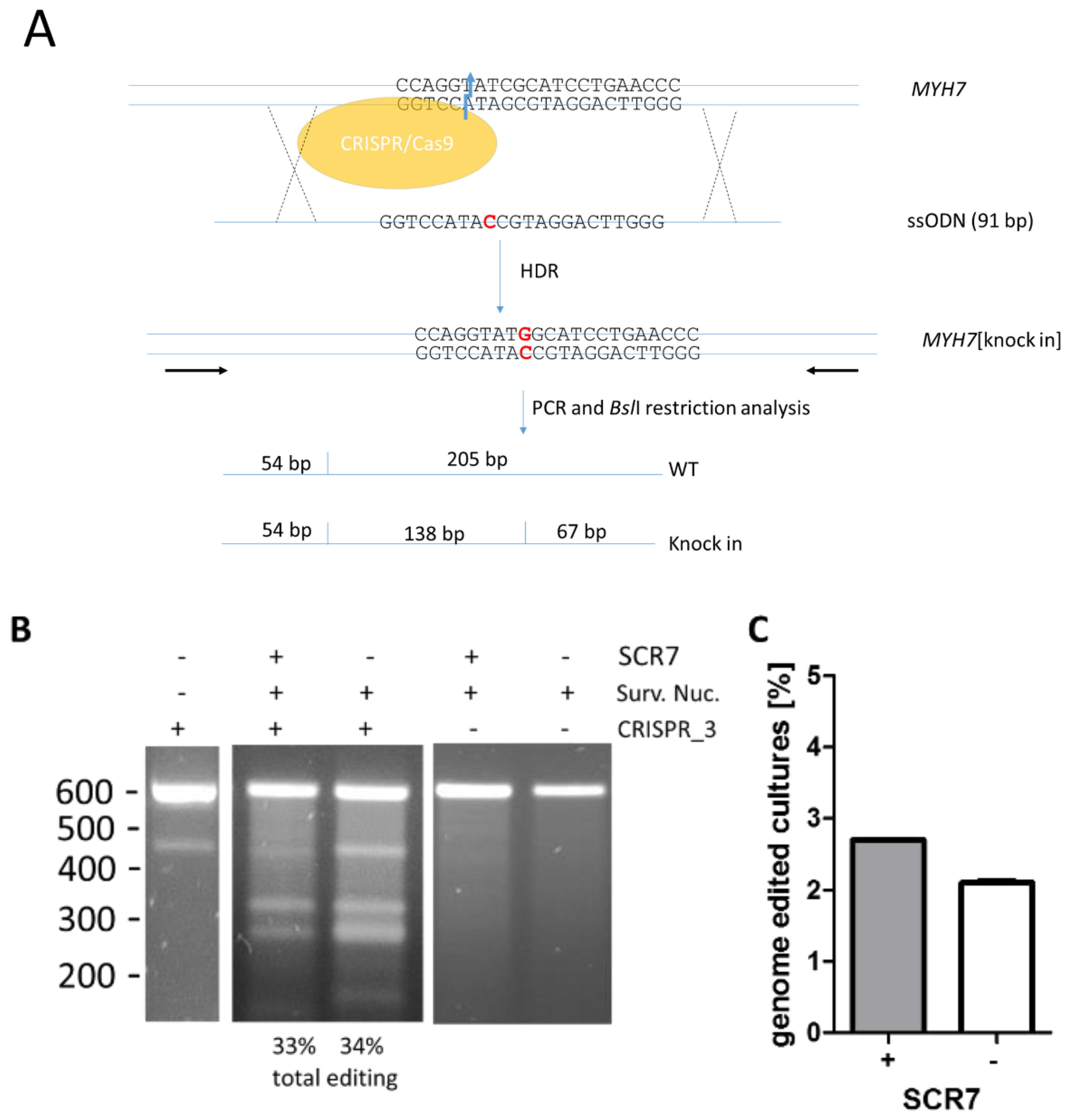
2.1. Targeting Plasmids and Donor DNA
2.2. Porcine Fetal Fibroblasts and Genome Editing
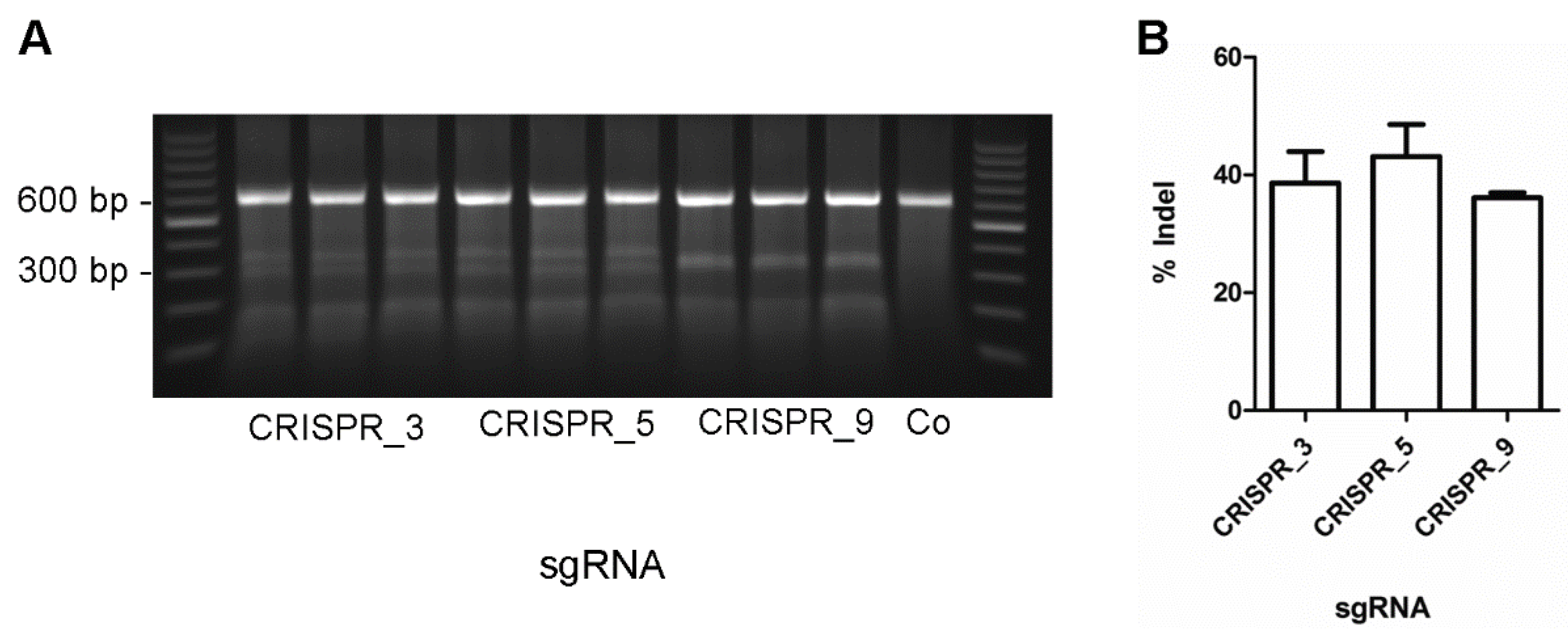
2.3. Surveyor Nuclease Assay
2.4. Screening for R723G-Genome Edited DNA
2.5. Sequence Analysis of R723G-Positive Cultures
2.6. Data Analysis
3. Results
3.1. Effects of SCR7
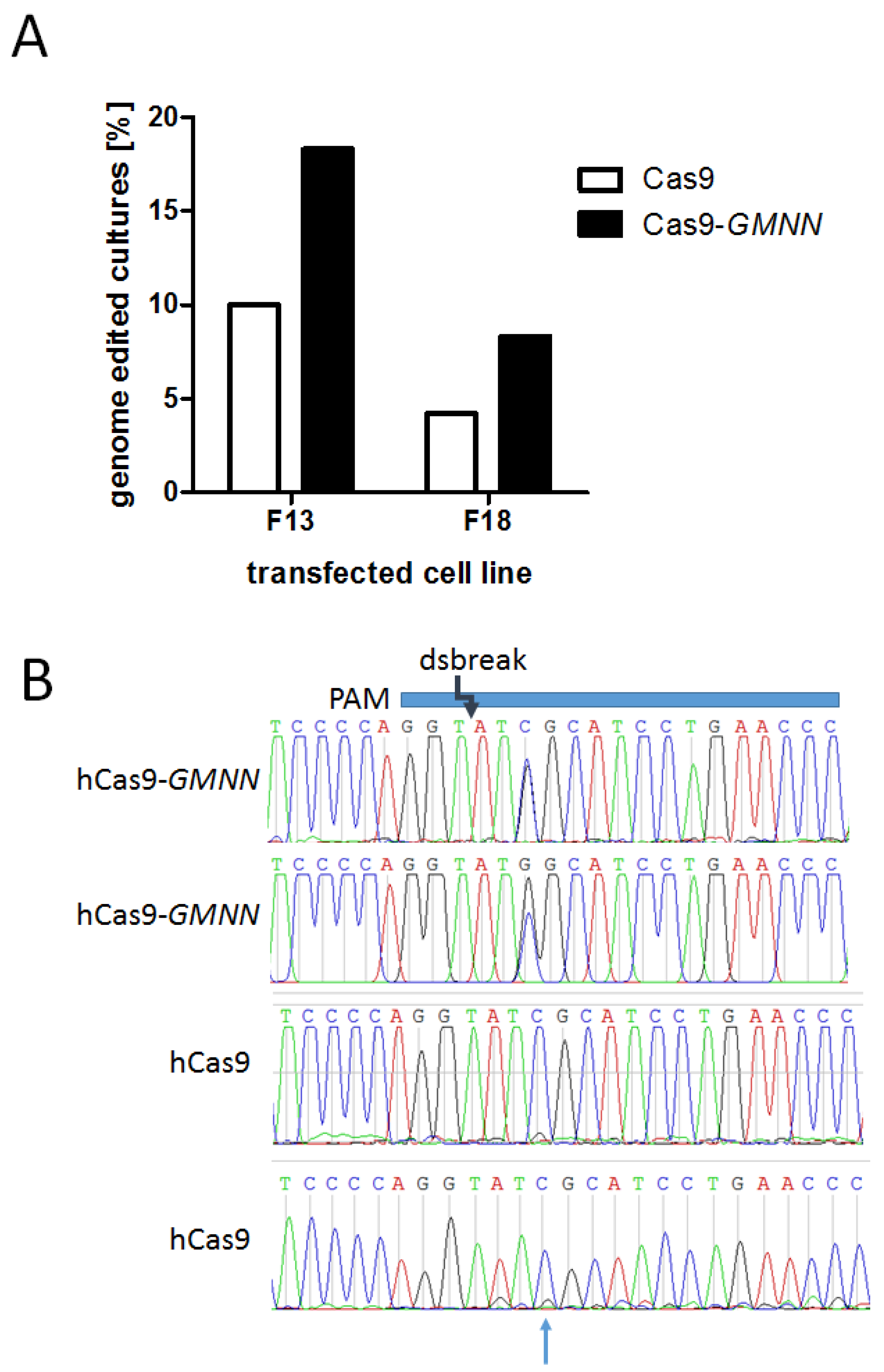
3.2. Effects of the Cas9-GMNN Fusion Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yao, J.; Huang, J.; Zhao, J. Genome editing revolutionize the creation of genetically modified pigs for modeling human diseases. Hum. Genet. 2016, 135, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Karvelis, T.; Gasiunas, G.; Siksnys, V. Programmable DNA cleavage in vitro by Cas9. Biochem. Soc. Trans. 2013, 41, 1401–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.-M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.J.; Hu, P.; Han, M.; Ellis, N.; Jasin, M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001, 15, 3237–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsk, K.; Kühn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, B.; Fan, Y.; He, W.; Zhu, D.; Niu, X.; Wang, D.; Ou, Z.; Luo, M.; Sun, X. Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev. 2015, 24, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, X.; Zhong, C.; Mo, J.; Quan, R.; Yang, J.; Liu, D.; Li, Z.; Yang, H.; Wu, Z. Small molecules enhance CRISPR/Cas9-mediated homology-directed genome editing in primary cells. Sci. Rep. 2017, 7, 8943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.F.; Chin, L. Post-translational regulation of Cas9 during G1 enhances homology-directed repair. Cell Rep. 2016, 14, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Howden, S.E.; McColl, B.; Glaser, A.; Vadolas, J.; Petrou, S.; Little, M.H.; Elefanty, A.G.; Stanley, E.G. A Cas9 variant for efficient generation of indel-free Knockin or gene-corrected human pluripotent stem cells. Stem Cell Rep. 2016, 7, 508–517. [Google Scholar] [CrossRef] [PubMed]
- McGarry, T.J.; Kirschner, M.W. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef]
- Pauklin, S.; Vallier, L. The cell-cycle state of stem cells determines cell fate propensity. Cell 2013, 155, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.; Ramackers, W.; Tiede, A.; Lucas-Hahn, A.; Herrmann, D.; Barg-Kues, B.; Schuettler, W.; Friedrich, L.; Schwinzer, R.; Winkler, M.; et al. Pigs transgenic for human thrombomodulin have elevated production of activated protein C. Xenotransplantation 2009, 16, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Montag, J.; Petersen, B.; Flögel, A.K.; Becker, E.; Lucas-Hahn, A.; Cost, G.J.; Mühlfeld, C.; Kraft, T.; Niemann, H.; Brenner, B. Successful knock-in of hypertrophic cardiomyopathy-mutation R723G into the MYH7 gene mimics HCM pathology in pigs. Sci. Rep. 2018, 8, 4786. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Berma, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauschild, J.; Petersen, B.; Queisser, Y.S.A.L.; Carnwath, J.W.; Lucas-Hahn, A.; Zhang, L.; Meng, X.; Gregory, P.D.; Schwinzerd, R.; Cost, G.J.; et al. Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc. Natl. Acad. Sci. USA 2011, 108, 12013–12017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Ouyang, H.; Xie, Z.; Yao, C.; Guo, N.; Li, M.; Jiao, H.; Pang, D. Efficient generation of myostatin mutations in pigs using the CRISPR/Cas9 System. Sci. Rep. 2015, 5, 16623. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Tang, X.; Xie, Z.; Zou, X.; Li, M.; Yuan, H.; Guo, N.; Ouyang, H.; Jiao, H.; Pang, D. CRISPR/Cas9-mediated knockout of myostatin in Chinese indigenous Erhualian pigs. Transgenic Res. 2017, 26, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chen, W.; Zhang, X.; Yu, L.; Dong, W.; Pan, S.; Gao, S.; Huang, X.; Zhang, L. Increasing the efficiency of CRISPR/Cas9-mediated precise genome editing in rats by inhibiting NHEJ and using Cas9 protein. RNA Biol. 2016, 13, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Meng, X.; Liu, Q.; Li, J.; Wang, K. Increasing the efficiency of CRISPR-Cas9-VQR precise genome editing in rice. Plant Biotechnol. J. 2018, 16, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khare, L.; Astrinidis, A.; Senapedis, W.; Adams, P.D.; Henske, E.P. Expression of wild type and mutant TSC2, but not TSC1, causes an increase in the G1 fraction of the cell cycle in HEK293 cells. J. Med. Genet. 2002, 39, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Pardee, A.B. G1 events and regulation of cell proliferation. Science 1989, 246, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, M.; Roberts, J.M. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science 1993, 259, 1908–1912. [Google Scholar] [CrossRef] [PubMed]
- Bialk, P.; Rivera-Torres, N.; Strouse, B.; Kmiec, E.B. Regulation of gene editing activity directed by single-stranded oligonucleotides and CRISPR/Cas9 systems. PLoS ONE 2015, 10, e0129308. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.D.; Ray, G.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| sgRNA | Sequence |
|---|---|
| 723_3 | AGGGTTCAGGATGCGATACCTGG |
| 723_5 | TGCGTGGCCTTAGATTCTGTGGG |
| 723_9 | GATTCACCCAAGTTCTGCATTGG |
| Fibroblasts | Cas9 | Cas9-GMNN | ||||
|---|---|---|---|---|---|---|
| Analyzed Cultures | Genome Edited Cultures | Analyzed Cultures | Genome Edited Cultures | |||
| Absolute | Percent | Absolute | Percent | |||
| PFF F13 | 180 | 18 | 10.0% | 180 | 33 | 18.3% |
| PFF F18 | 95 | 4 | 4.2% | 84 | 7 | 8.3% |
| Total | 275 | 22 | 8.0% | 264 | 40 | 15.2% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerlach, M.; Kraft, T.; Brenner, B.; Petersen, B.; Niemann, H.; Montag, J. Efficient Knock-in of a Point Mutation in Porcine Fibroblasts Using the CRISPR/Cas9-GMNN Fusion Gene. Genes 2018, 9, 296. https://doi.org/10.3390/genes9060296
Gerlach M, Kraft T, Brenner B, Petersen B, Niemann H, Montag J. Efficient Knock-in of a Point Mutation in Porcine Fibroblasts Using the CRISPR/Cas9-GMNN Fusion Gene. Genes. 2018; 9(6):296. https://doi.org/10.3390/genes9060296
Chicago/Turabian StyleGerlach, Max, Theresia Kraft, Bernhard Brenner, Björn Petersen, Heiner Niemann, and Judith Montag. 2018. "Efficient Knock-in of a Point Mutation in Porcine Fibroblasts Using the CRISPR/Cas9-GMNN Fusion Gene" Genes 9, no. 6: 296. https://doi.org/10.3390/genes9060296
APA StyleGerlach, M., Kraft, T., Brenner, B., Petersen, B., Niemann, H., & Montag, J. (2018). Efficient Knock-in of a Point Mutation in Porcine Fibroblasts Using the CRISPR/Cas9-GMNN Fusion Gene. Genes, 9(6), 296. https://doi.org/10.3390/genes9060296