Genome-Wide Identification and Functional Prediction of Novel Drought-Responsive lncRNAs in Pyrus betulifolia

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Dehydration Treatment
2.2. RNA Extraction, Library Construction, and lncRNA Sequencing
2.3. Data Analysis
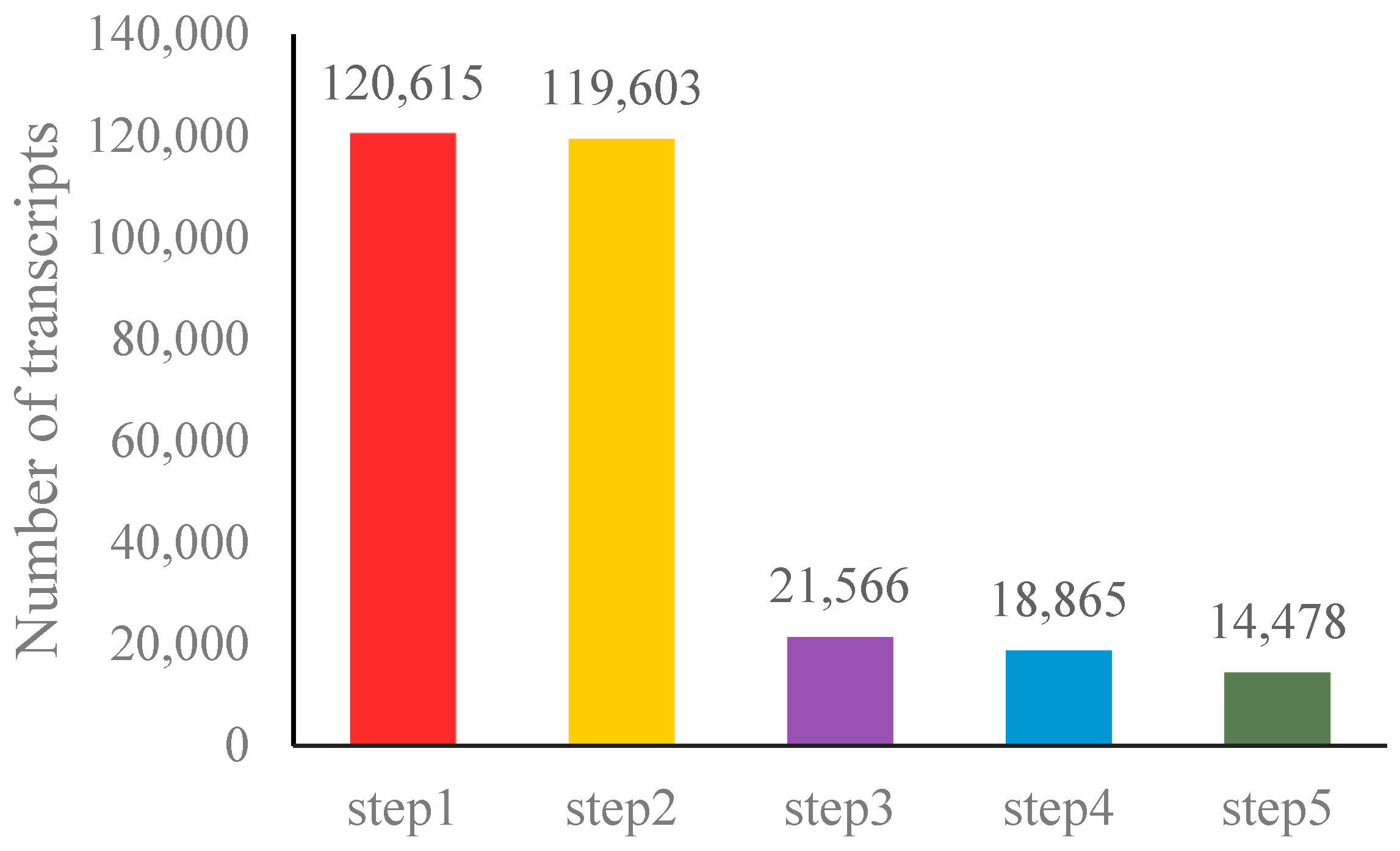
2.3.1. Quality Control, Transcriptome Assembly and lncRNAs Identification
2.3.2. Differential Expression Analysis
2.3.3. Putative Target Gene Prediction and Enrichment Analysis
2.3.4. Quantitative Real-Time Polymerase Chain Reaction Analysis
3. Results
3.1. Identification of lncRNAs in the Birch-Leaf Pear
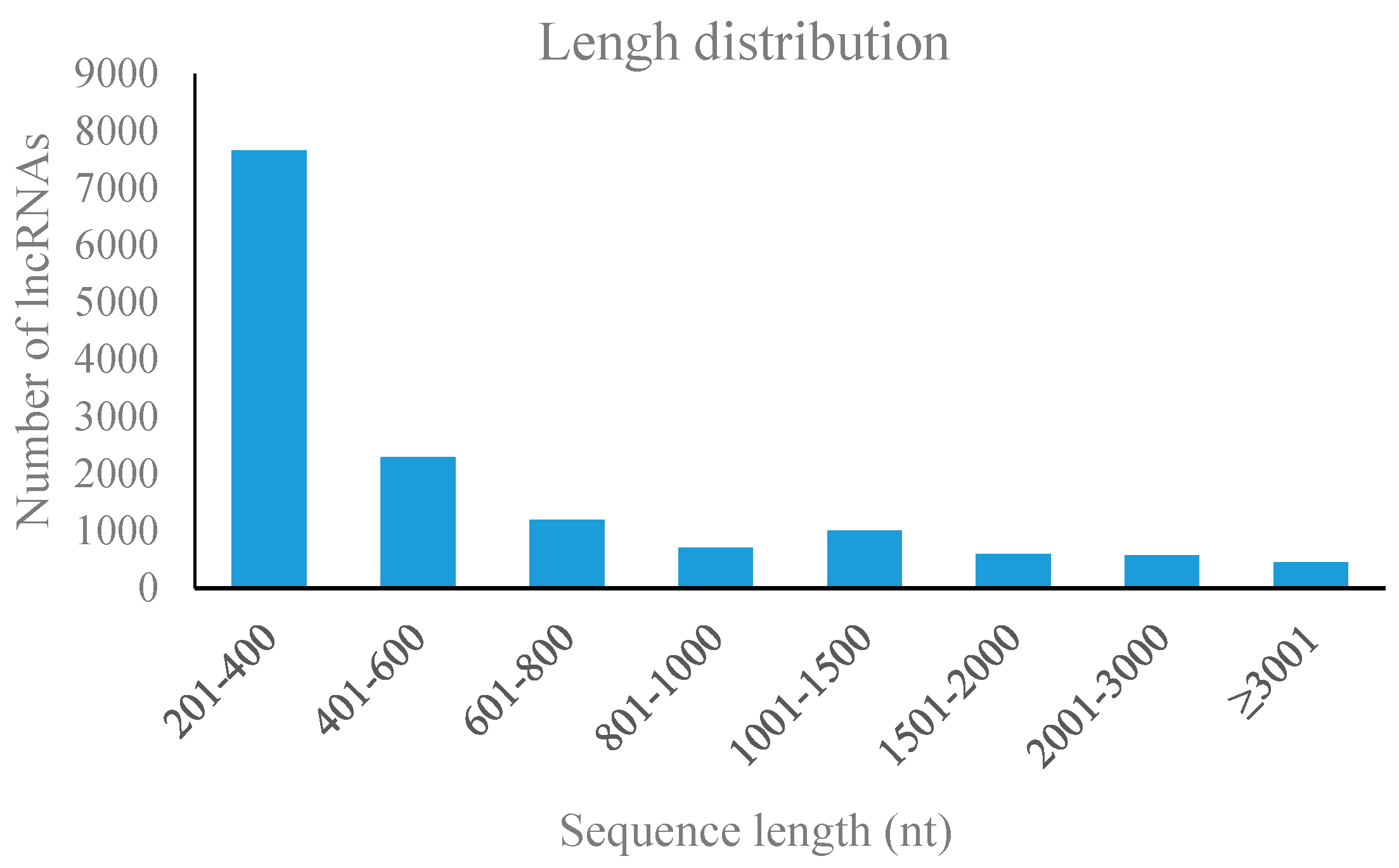
3.2. Characterization of Pear lncRNAs
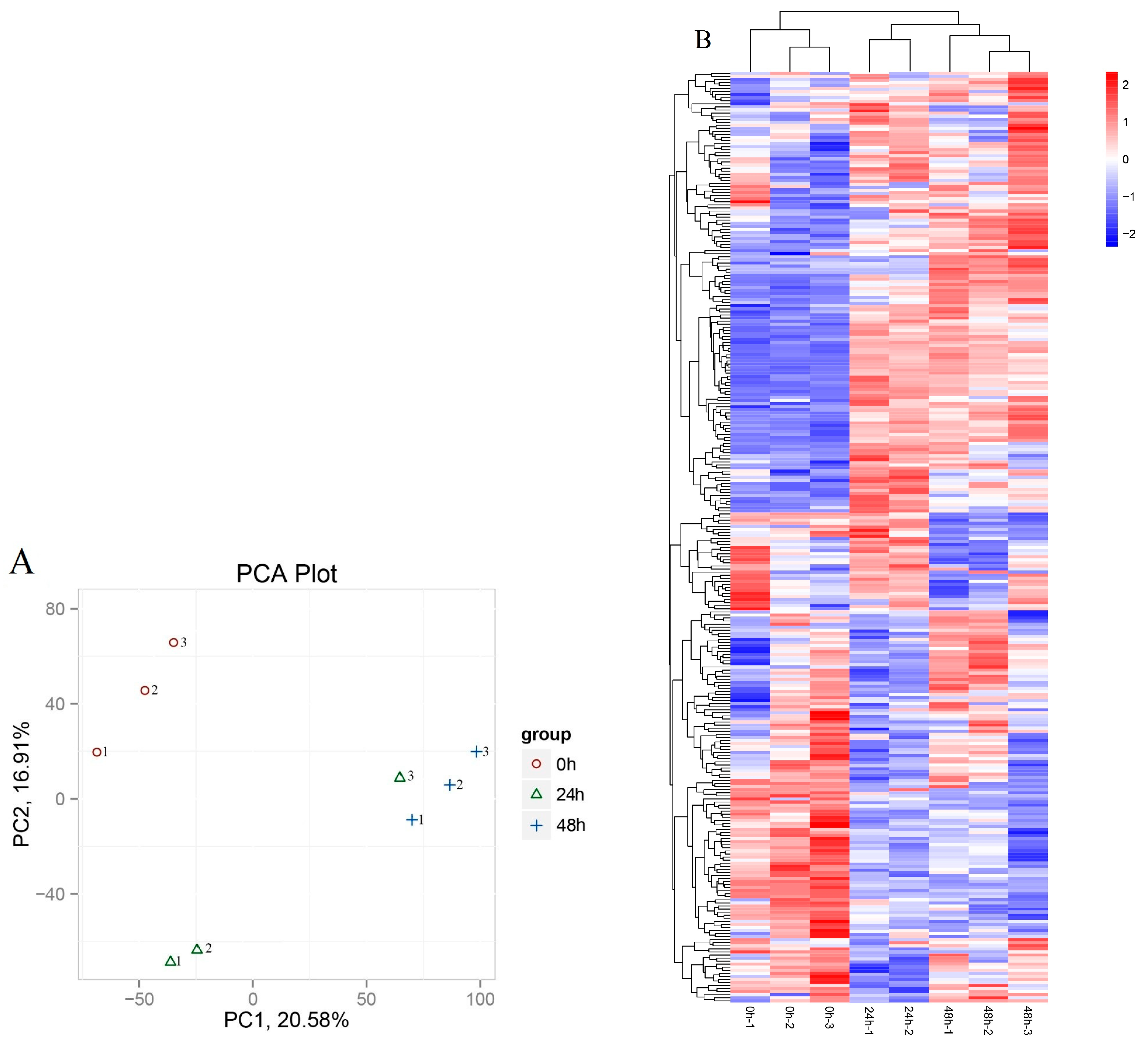
3.3. Identification of lncRNAs that Respond to Drought-Stress Treatments
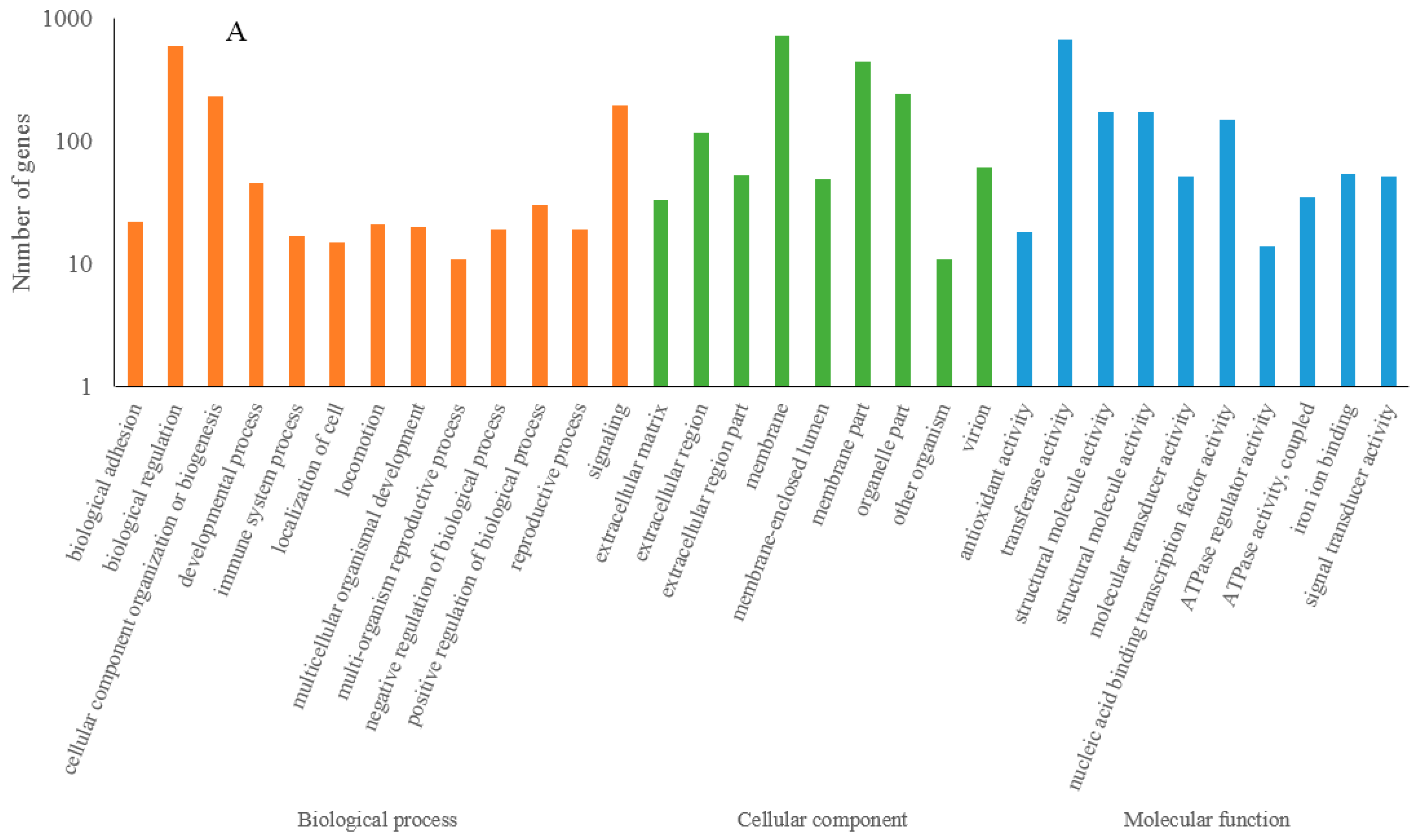
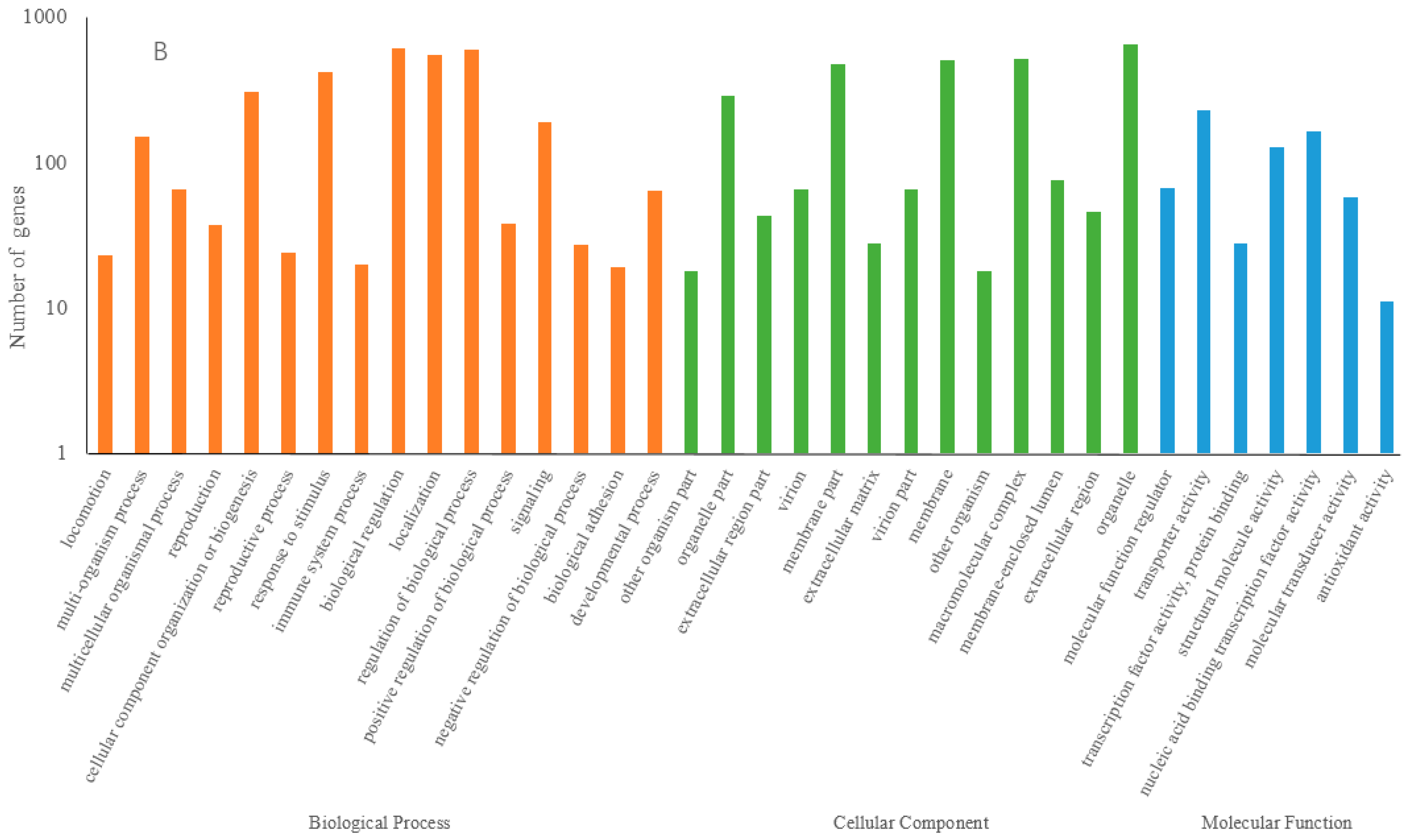
3.4. Functional Prediction of Drought Response lncRNAs
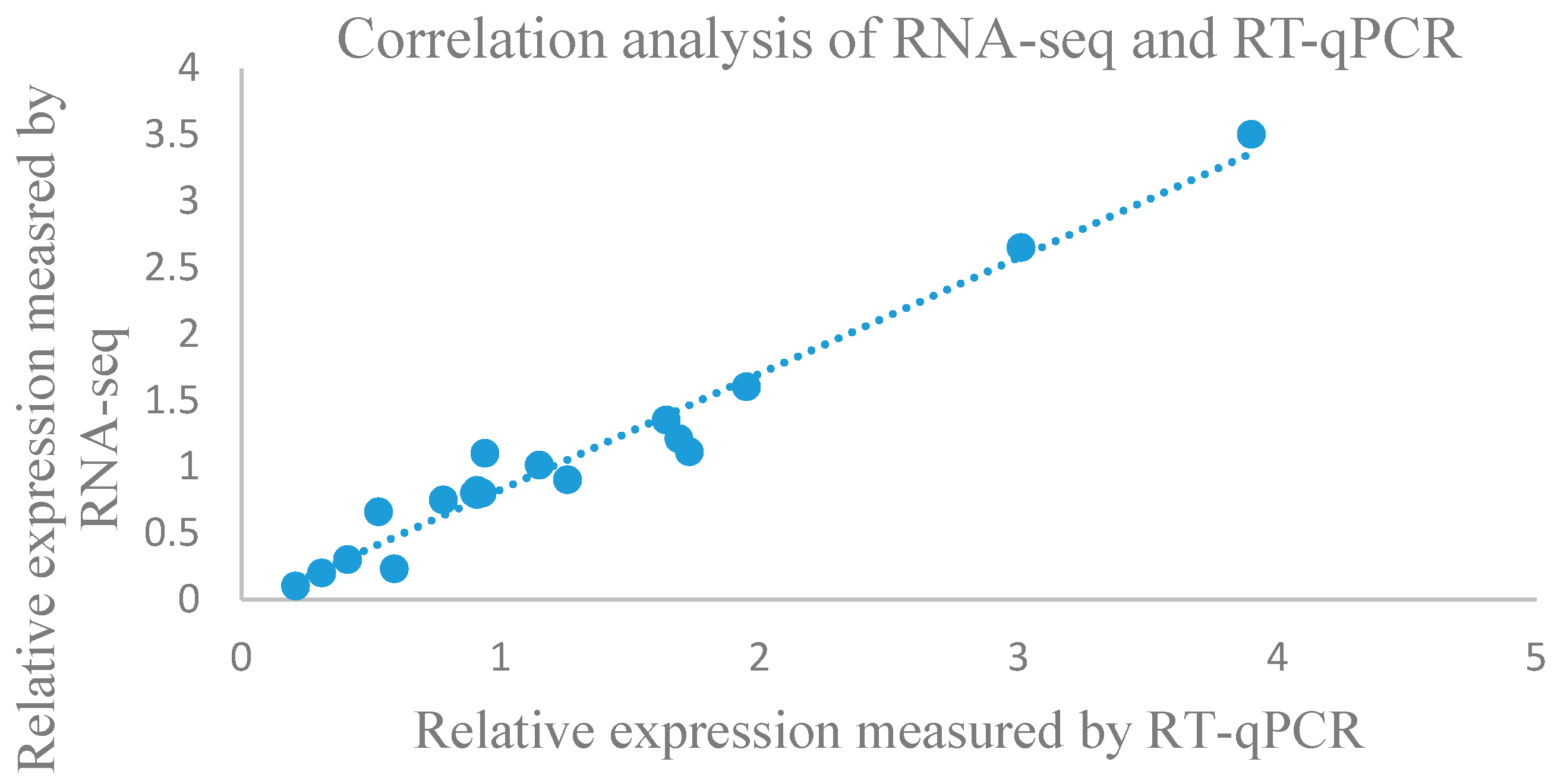
3.5. Real-Time Quantitative Polymerase Chain Reaction Validation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wu, J.; Wang, Z.; Shi, Z.; Zhang, S.; Ming, R.; Zhu, S.; Khan, M.A.; Tao, S.; Korban, S.S.; Wang, H.; et al. The genome of the pear (Pyrus bretschneideri Rehd). Genome Res. 2013, 23, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Kan, J.; Liu, T.; Ma, N.; Li, H.; Li, X.; Wang, J.; Zhang, B.; Chang, Y.; Lin, J. Transcriptome analysis of Callery pear (Pyrus calleryana) reveals a comprehensive signalling network in response to Alternaria alternata. PLoS ONE 2017, 12, e0184988. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Dong, Q.; Li, C.; Liu, C.; Ma, F. Genome Wide Identification and Characterization of Apple bHLH Transcription Factors and Expression Analysis in Response to Drought and Salt Stress. Front. Plant Sci. 2017, 8, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, C.L.; Baldo, A.M.; Moore, J.T.; Jenkins, R.M.; Soffe, D.S.; Wisniewski, M.E.; Norelli, J.L.; Farrell, R.E. Genes responding to water deficit in apple (Malus × domestica Borkh.) roots. BMC Plant Biol. 2014, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Guo, X.; Zhao, D.; Zhang, Q.; Jiang, Y.; Wang, Y.; Peng, X.; Wei, Y.; Zhai, Z.; Zhao, W.; et al. Cloning and expression profiling of the PacSnRK2 and PacPP2C gene families during fruit development, ABA treatment, and dehydration stress in sweet cherry. Plant Physiol. Biochem. 2017, 119, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.R.; Ergül, A.; Grimplet, J.; Tillett, R.L.; Tattersall, E.A.; Bohlman, M.C.; Vincent, D.; Sonderegger, J.; Evans, J.; Osborne, C.; et al. Water and salinity stress in grapevines: Early and late changes in transcript and metabolite profiles. Funct. Integr. Genom. 2007, 7, 111–134. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Hur, Y.Y.; Jung, S.M.; Choi, Y.J.; Nam, J.C.; Park, J.G.; Koh, S.W. Transcript profiling of native Korean grapevine species Vitis flexuosa, exposed to dehydration and rehydration treatment. Hortic. Environ. Biotechnol. 2017, 58, 66–77. [Google Scholar] [CrossRef]
- Shen, J.; Xiao, Q.; Qiu, H.; Chen, C.; Chen, H. Integrative effect of drought and low temperature on litchi (Litchi chinensis Sonn.) floral initiation revealed by dynamic genome-wide transcriptome analysis. Sci. Rep. 2016, 6, 32005. [Google Scholar] [CrossRef] [PubMed]
- Li, K.Q.; Xu, X.Y.; Huang, X.S. Identification of Differentially Expressed Genes Related to Dehydration Resistance in a Highly Drought-Tolerant Pear, Pyrus betulaefolia, as through RNA-Seq. PLoS ONE 2016, 11, e0149352. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223. [Google Scholar] [PubMed]
- Liu, J.; Wang, H.; Chua, N.H. Long noncoding RNA transcriptome of plants. Plant Biotechnol. J. 2015, 13, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, J. The Long-Noncoding RNA ELENA1 Functions in Plant Immunity. Plant Cell 2017, 29, 916. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Lin, Y.; Wu, J. Long non-coding RNA expression profiling of mouse testis during postnatal development. PLoS ONE 2013, 8, e75750. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Wang, Y.; Yao, Y.; Song, N.; Hu, Z.; Qin, D.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, X.; Wang, C.; Xu, Z.; Wang, Y.; Liu, X.; Kang, Z.; Ji, W. Long non-coding genes implicated in response to stripe rust pathogen stress in wheat (Triticum aestivum, L.). Mol. Biol. Rep. 2013, 40, 6245–6253. [Google Scholar] [CrossRef] [PubMed]
- Jalali, S.; Bhartiya, D.; Lalwani, M.K.; Sivasubbu, S.; Scaria, V. Systematic transcriptome wide analysis of lncRNA-miRNA Interactions. PLoS ONE 2013, 8, e53823. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, C.; Bao, H.; Chen, H.; Wang, Y. Genome-wide identification and characterization of novel lncRNAs in Populus, under nitrogen deficiency. Mol. Genet. Genom. 2016, 291, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Han, Z.; Guo, Q.; Liu, Y.; Zheng, Y.; Wu, F.; Jin, W. Identification of maize long non-coding RNAs responsive to drought stress. PLoS ONE 2014, 9, e98958. [Google Scholar] [CrossRef] [PubMed]
- Marker, C.; Zemann, A.; Terhörst, T.; Kiefmann, M.; Kastenmayer, J.P.; Green, P.; Bachellerie, J.P.; Brosius, J.; Hüttenhofer, A. Experimental RNomics: Identification of 140 candidates for small non-messenger RNAs in the plant Arabidopsis thaliana. Curr. Biol. 2002, 12, 2002–2013. [Google Scholar] [CrossRef]
- Li, L.; Wang, X.; Sasidharan, R.; Stolc, V.; Deng, W.; He, H.; Korbel, J.; Chen, X.; Tongprasit, W.; Ronald, P.; et al. Global identification and characterization of transcriptionally active regions in the rice genome. PLoS ONE 2007, 2, e294. [Google Scholar] [CrossRef] [PubMed]
- Amor, B.B.; Wirth, S.; Merchan, F.; Laporte, P.; d’Aubenton-Carafa, Y.; Hirsch, J.; Maizel, A.; Mallory, A.; Lucas, A.; Deragon, J.M.; et al. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2009, 19, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Parker, B.J.; Weiller, G.F. In Silico identification and characterization of mRNA-like noncoding transcripts in Medicago truncatula. Silico Biol. 2007, 7, 485–505. [Google Scholar]
- Ren, H.; Wang, G.; Chen, L.; Jiang, J.; Liu, L.; Li, N.; Zhao, J.; Sun, X.; Zhou, P. Genome-wide analysis of long non-coding RNAs at early stage of skin pigmentation in goats (Capra hircus). BMC Genom. 2016, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Chen, Y.Q. Long noncoding RNAs: New regulators in plant development. Biochem. Biophys. Res. Commun. 2013, 436, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Boerner, S.; McGinnis, K.M. Computational identification and functional predictions of long noncoding RNA in Zea mays. PLoS ONE 2012, 7, e43047. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E.; et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, Y.; Sun, P.; Liu, J.; Yue, X.; Niu, Q.; Teng, Y. Chloroplast DNA-based genetic diversity and phylogeography of Pyrus betulaefolia, (Rosaceae) in Northern China. Tree Genet. Genomes 2014, 10, 739–749. [Google Scholar] [CrossRef]
- Wang, H.; Lin, J.; Li, X.G.; Chang, Y. Genome-wide identification of pear HD-Zip gene family and expression patterns under stress induced by drought, salinity, and pathogen. Acta Physiol. Plant. 2015, 37, 189. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Lin, J.; Wang, Z.; Yang, Q.; Chang, Y. Transcriptome sequencing and analysis of major genes involved in calcium signaling pathways in pear plants (Pyrus calleryana Decne.). BMC Genom. 2015, 16, 738. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protocols. 2012, 7, 562. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunesekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar]
- Lin, M.F.; Jungreis, I.; Kellis, M. PhyloCSF: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011, 27, i275–i282. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Guo, W.; Liu, Y.; Wei, H.; Yang, S. Transcription analysis of cochlear development in minipigs. Acta Oto-Laryngol. 2017, 1. [Google Scholar] [CrossRef]
- Lobo, I. Basic Local Alignment Search Tool (BLAST). J. Mol. Biol. 2008, 215, 403–410. [Google Scholar]
- Bateman, A.; Birney, E.; Cerruti, L.; Durbin, R.; Griffiths-Jones, S.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2002, 28, 263–266. [Google Scholar] [CrossRef]
- Nawrocki, E.P. Annotating Functional RNAs in Genomes Using Infernal. In RNA Sequence, Structure, and Function: Computational and Bioinformatic Methods; Humana Press: New York, NY, USA, 2014; pp. 163–197. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple hypothesis testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Xu, M.; Shi, H.; Gao, X.; Liang, P. Genome-wide identification of lncRNAs associated with chlorantraniliprole resistance in diamondback moth Plutella xylostella (L.). BMC Genom. 2017, 18, 380. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Abbas, M.; Wen, Y.; Niu, D.; Wang, L.; Sun, Y.; Li, Y. Selection and validation of reference genes for quantitative gene expression analyses in black locust (Robinia pseudoacacia L.) using real-time quantitative PCR. PLoS ONE 2018, 13, e0193076. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCT method. Method 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, H.; Li, X.; Lin, J.; Wang, Z.; Yang, Q.; Chang, Y. Systematic selection and validation of appropriate reference genes for gene expression studies by quantitative real-time PCR in pear. Acta Physiol. Plant. 2015, 37, 40. [Google Scholar] [CrossRef]
- Lu, X.; Wang, X.; Chen, X.; Shu, N.; Wang, J.; Wang, D.; Wang, S.; Fan, W.; Guo, L.; Guo, X.; et al. Single-base resolution methylomes of upland cotton (Gossypium hirsutum L.) reveal epigenome modifications in response to drought stress. BMC Genom. 2017, 18, 297. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen, X.; Mu, M.; Wang, J.; Wang, X.; Wang, D.; Yin, Z.; Fan, W.; Wang, S.; Guo, L.; et al. Genome-wide analysis of long noncoding RNAs and their responses to drought stress in cotton (Gossypium hirsutum L.). PLoS ONE 2016, 11, e0156723. [Google Scholar] [CrossRef] [PubMed]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Di, C.; Yuan, J.; Wu, Y.; Li, J.; Lin, H.; Hu, L.; Zhang, T.; Qi, Y.; Gerstein, M.B.; Guo, Y.; et al. Characterization of stress-responsive lncRNAs in Arabidopsis thaliana by integrating expression, epigenetic and structural features. Plant J. 2014, 80, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Lin, J.; Li, H.; Li, X.; Yang, Q.; Cheng, Z.M.; Chang, Y. Characterization of CIPK Family in Asian Pear (Pyrus bretschneideri Rehd) and Co-expression Analysis Related to Salt and Osmotic Stress Responses. Front. Plant Sci. 2016, 7, 1361. [Google Scholar] [CrossRef] [PubMed]
- Pastori, G.M.; Foyer, C.H. Common components, networks, and pathways of cross-tolerance to stress. The central role of “redox” and abscisic acid-mediated controls. Plant Physiol. 2002, 129, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Seifert, S.; Lübbe, T.; Leuschner, C.; Finkeldey, R. De novo transcriptome assembly and analysis of differential gene expression in response to drought in European beech. PLoS ONE 2017, 12, e0184167. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Wu, B.; Zheng, H.; Hu, D.; Wang, X.; Duan, H.; Sun, Y.; Wang, J.; Zhang, Y.; Li, Y. Global Reprogramming of Transcription in Chinese Fir (Cunninghamia lanceolata) during Progressive Drought Stress and after Rewatering. Int. J. Mol. Sci. 2015, 16, 15194–15219. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Reads | Clean Reads | Clean Bases | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|
| h0–1 | 104,464,354 | 101,335,652 | 15.2 G | 0.01 | 97.61 | 93.56 | 43.19 |
| h0–2 | 92,734,618 | 90,096,636 | 13.51 | 0.02 | 95.63 | 88.03 | 44.27 |
| h0–3 | 89,700,722 | 87,193,746 | 13.08 G | 0.03 | 95.00 | 86.72 | 43.49 |
| h24–1 | 99,690,832 | 96,780,130 | 14.52 G | 0.02 | 97.51 | 93.32 | 43.07 |
| h24–2 | 96,641,070 | 94,021,570 | 14.1 G | 0.02 | 97.37 | 93.04 | 43.60 |
| h24–3 | 99,260,444 | 96,754,238 | 14.51 G | 0.02 | 97.57 | 93.46 | 43.59 |
| h48–1 | 102,684,392 | 99,947,264 | 14.99 G | 0.02 | 97.57 | 93.48 | 43.02 |
| h48–2 | 98,612,476 | 95,718,804 | 14.36 G | 0.02 | 97.56 | 93.45 | 43.60 |
| h48–3 | 95,021,280 | 92,290,956 | 13.84 G | 0.02 | 97.47 | 93.24 | 43.35 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Lin, J.; Kan, J.; Wang, H.; Li, X.; Yang, Q.; Li, H.; Chang, Y. Genome-Wide Identification and Functional Prediction of Novel Drought-Responsive lncRNAs in Pyrus betulifolia. Genes 2018, 9, 311. https://doi.org/10.3390/genes9060311
Wang J, Lin J, Kan J, Wang H, Li X, Yang Q, Li H, Chang Y. Genome-Wide Identification and Functional Prediction of Novel Drought-Responsive lncRNAs in Pyrus betulifolia. Genes. 2018; 9(6):311. https://doi.org/10.3390/genes9060311
Chicago/Turabian StyleWang, Jinxing, Jing Lin, Jialiang Kan, Hong Wang, Xiaogang Li, Qingsong Yang, Hui Li, and Youhong Chang. 2018. "Genome-Wide Identification and Functional Prediction of Novel Drought-Responsive lncRNAs in Pyrus betulifolia" Genes 9, no. 6: 311. https://doi.org/10.3390/genes9060311
APA StyleWang, J., Lin, J., Kan, J., Wang, H., Li, X., Yang, Q., Li, H., & Chang, Y. (2018). Genome-Wide Identification and Functional Prediction of Novel Drought-Responsive lncRNAs in Pyrus betulifolia. Genes, 9(6), 311. https://doi.org/10.3390/genes9060311