1. Introduction
Mammalian HTRA2 is a nuclear-encoded mitochondrial serine protease which is expressed as a 49 kDa precursor protein that is cleaved to a 38 kDa mature protein upon import into the mitochondria [
1]. It includes a mitochondrial targeting sequence, N-terminal transmembrane domain, a serine protease domain and a C-terminal PDZ protein interaction domain [
2], named after the first three proteins found to contain this domain: Post synaptic density protein (PSD95),
Drosophila discs large 1 tumor suppressor (dlg1), and Zonula occludens-1 protein (ZO-1). Under normal conditions, HTRA2 is localized to the mitochondria, but is released to the cytosol after apoptotic stimuli such as UV irradiation and oxidative stress. In the cytosol, it initiates caspase-dependent programmed cell death as well as cell death by caspase-independent (proteolysis) pathways [
1,
3,
4,
5].
Mutations in either the serine protease or PDZ domains are reported to be associated with autosomal dominant Parkinson’s disease (PD) [
6,
7,
8]. HTRA2 was also found to colocalize with another PD-linked protein, α-synuclein, in Lewy Bodies, neuronal protein aggregates that are the hallmark of most forms of PD [
9]. The role of HTRA2 in PD pathogenesis is thought to result from loss of its ability to remove misfolded or damaged mitochondrial proteins through its serine protease function. Mutations to this function result in the accumulation and aggregation of proteins, which is characteristic of PD. HTRA2 also interacts with other PD proteins such as PINK1, which has been reported to phosphorylate HTRA2 and this phosphorylation at S142 increases its proteolytic activity [
10,
11,
12,
13,
14]. Amino acid substitutions (A141S, G399S) close to this and another phosphorylation site (S400) have been implicated in PD and are believed to act by inhibiting phosphorylation and enzyme activity [
13].
Conflicting data about the role of HTRA2 in PD has also been reported [
2]. For example, Simon-Sanchez and Singleton [
15] found previously reported pathogenic HTRA2 mutations in neurologically normal controls, while others found no strong association between HTRA2 variants and PD in population genetic studies [
16,
17,
18]. The discrepancies between the negative population studies and the positive studies on PD kindreds carrying HTRA2 mutations may arise from combinations of low penetrance, allele rarity and genetic modifiers.
Regardless of its place in the pantheon of PD-associated proteins, HTRA2 is a mitochondrial protein whose genetic loss of function thus represents a mitochondrial disease. However, there has also been disagreement about the cytopathological effects of HTRA2 loss on the mitochondria, particularly in relation to its role in the Parkin-PINK1 pathway, which affects mitochondrial integrity and dynamics [
2,
19]. In animals and humans, the complexities of mitochondrial biology are such that the genotype-phenotype nexus is broken—the same mitochondrial defects can produce dramatically different outcomes in different individuals, while different mutations affecting different genes can produce clinically similar consequences. These complexities are bypassed in the simple mitochondrial disease model,
Dictyostelium discoideum [
20].
D. discoideum is one of 10 valuable non-mammalian models according to the USA National Institutes of Health [
20,
21]. Techniques such as genetic transformation [
22], determination of gene copy numbers [
23], gene knockdown by antisense RNA inhibition [
24], GFP (green fluorescent protein) tagging of proteins, protein extraction, purification and immunodetection [
25] and accessible measurement of diverse phenotypes [
26] are well developed. This provides a readily exploited opportunity to manipulate HTRA2 expression levels at the molecular level and examine the consequences in vivo in
D. discoideum.
Although the serine protease activity of HTRA2 has been reported to play a positive protective role in removing aberrant proteins in the mitochondria, it is unclear what level of the serine protease activity should be maintained in this process. Would elevated expression and activity of HTRA2 in the mitochondria cause any damage to cells? In a recently reported PD case, a P143A substitution in HTRA2 resulted in hyperphosphorylation (and presumably elevated activity) and was suggested to have contributed to PD pathology and mitochondrial dysfunction [
27]. What would be the consequences if the serine protease activity of mitochondrial HTRA2 is removed? We confirm here that HTRA2 in
Dictyostelium is localized in the mitochondria, from which location it plays a positive and/or protective role in regulating growth and development. Its knockdown causes phenotypic outcomes that are only partly reminiscent of other mitochondrial defects in this organism and are also distinct from those recently reported to result from loss of another PD-associated protein DJ-1 [
28]. Despite its positive, cytoprotective roles, mitochondrial overexpression of wild type HTRA2 is lethal because of its proteolytic activity. Overexpression of a protease-dead mutant form is not lethal but faithfully phenocopies the effects of reduced expression. Mitochondrial respiration was not impaired either by HTRA2 knockdown or overexpression of protease-dead HTRA2.
3. Results
3.1. A Single Homologue of Human HTRA2 Is Encoded in the Dictyostelium discoideum Genome and Localized in the Mitochondria
A BLAST (basic local alignment search tool) search at dictyBase [
32] using the protein sequence of human HTRA2 [
33] as the query sequence identified a single homologous protein (which we designate HTRA2) encoded in the
D. discoideum genome by a gene (accession number DDB_G0281081) we designate as
htrA, full protein sequence alignment shown in (
Figure 1).
Human HTRA2 is reported to be localized in the mitochondria [
37] and, to verify this predicted localization in
D. discoideum, three mitochondrial prediction programs (MitoProt II [
35], Predotar [
38] and Helical Wheel [
39]) were used to determine the likelihood of HTRA2’s mitochondrial localization. The results predict that, like its human counterpart, HTRA2 is localized to the mitochondria in
D. discoideum (
Figure 1,
Figure S1,
Table S1,
Figure S2). As well as the predicted mitochondrial targeting signal at the N-terminus of the protein sequence, InterProScan analysis [
36] detected a serine protease domain (residues 141–345) and PDZ protein-binding domain (residues 352–453) in the locations expected from the positions of the corresponding regions in the human protein (
Figure 1).
Our attempts to confirm experimentally the mitochondrial localization of
Dictyostelium HTRA2 by expressing wild type HTRA2 tagged at the C-terminus were unsuccessful (
Section 3.5.1) and we tried but were unable to generate an anti-HTRA2 antibody that could be used in immunofluorescence microscopy. However, we did succeed in isolating transformants expressing a C-terminally GFP-tagged, protease-dead mutant (S
300 substituted with alanine) form of the protein (
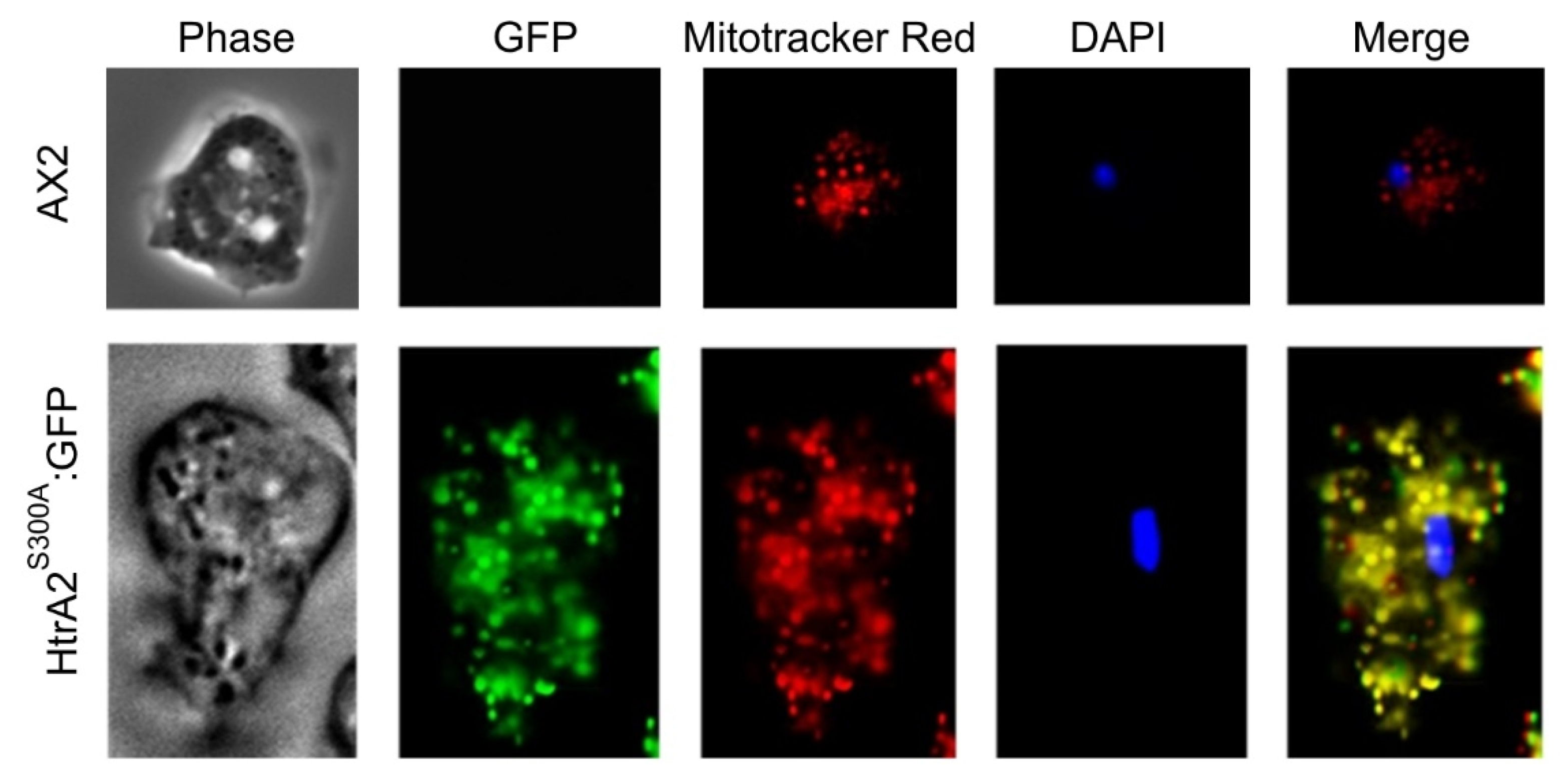
Section 3.5.2). Epifluorescence microscopy revealed the fusion protein to be located in the mitochondria as expected (
Figure 2). C-terminal GFP-tagging is a standard approach to tagging mitochondrial proteins because the mitochondrial targeting signal is normally (as in this case) a leader peptide at the N-terminus.
3.2. Creation of HTRA2 Knockdown Transformants
To study the phenotypic outcomes of loss of HTRA2 function in
Dictyostelium, we created a HTRA2 antisense-inhibition construct (pPROF689) and used it to isolate multiple knockdown transformants of the parental
D. discoideum strain AX2. The constructs insert randomly into the genome in a process accompanied by rolling circle replication which generates transformants, each with a unique number of copies of the construct so that each transformant has a different level of antisense inhibition [
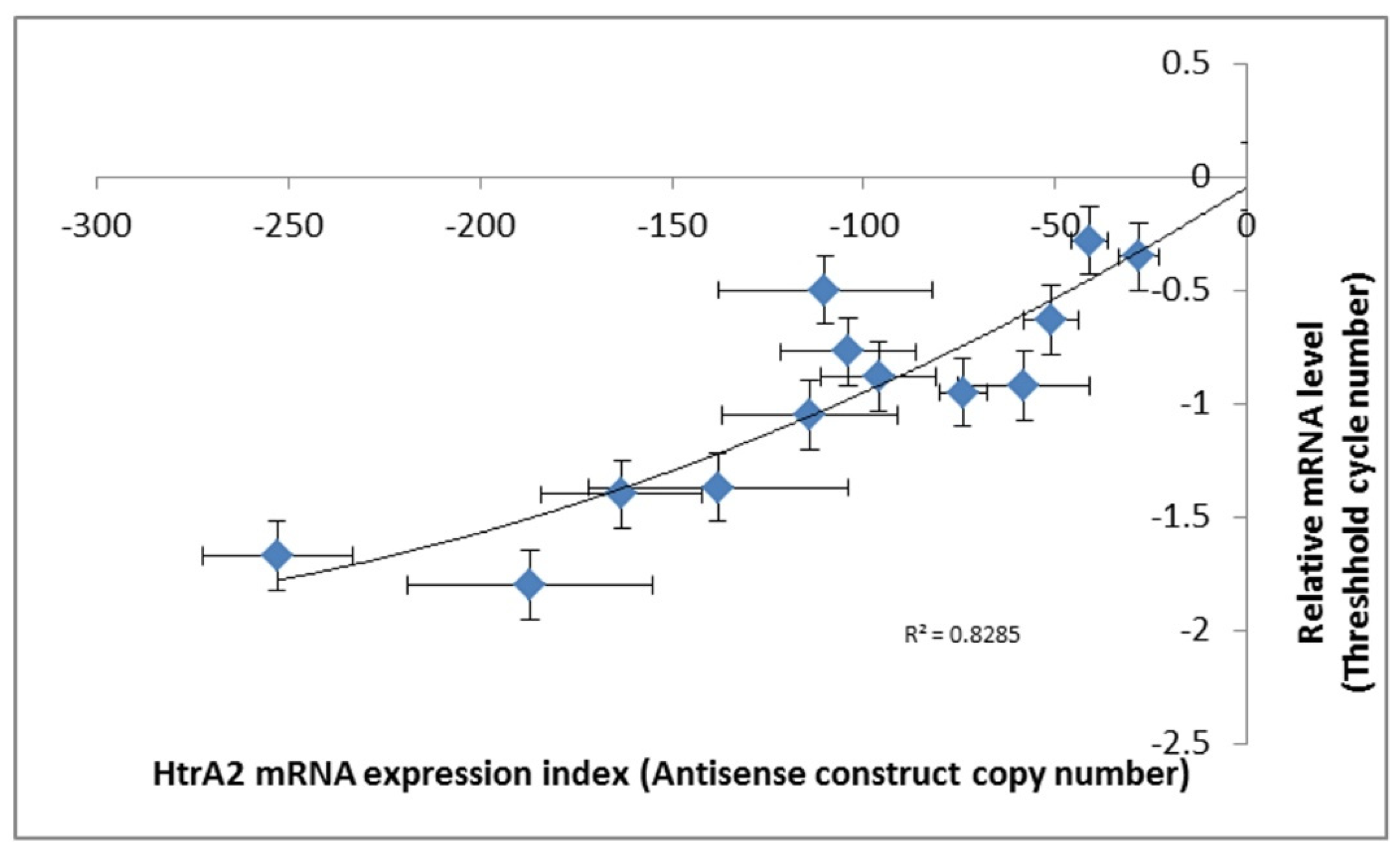
40]. The copy numbers of the inserted construct were determined by qPCR and the expression levels of the construct were measured by qRT-PCR. The relationship between
htrA mRNA expression levels and the copy numbers of HTRA2 antisense construct are shown in (
Figure 3).
3.3. Mitochondrial HTRA2 Is Needed for Normal Morphogenesis and Growth, but Not for Phototaxis
In
D. discoideum, mitochondrial respiratory dysfunction has been created by various genetic methods, and in each case the mitochondrially diseased strains presented consistent phenotypes, including impaired growth, phototaxis and morphology [
30,
41,
42,
43,
44]. These included heteroplasmic knockout of nine different genes in the mitochondrial genome, knockout of a nuclear-encoded Complex I assembly factor (MidA) and knockdown of the nuclear-encoded mitochondrial chaperone (Chaperonin 60) required for folding of mitochondrial proteins [
20]. If inhibition of HTRA2 causes mitochondrial respiratory dysfunction, it should generate the same phenotypes caused by
D. discoideum mitochondrial respiratory disease. To verify this, the
D. discoideum transformants containing HTRA2 antisense-inhibition construct were characterized phenotypically (
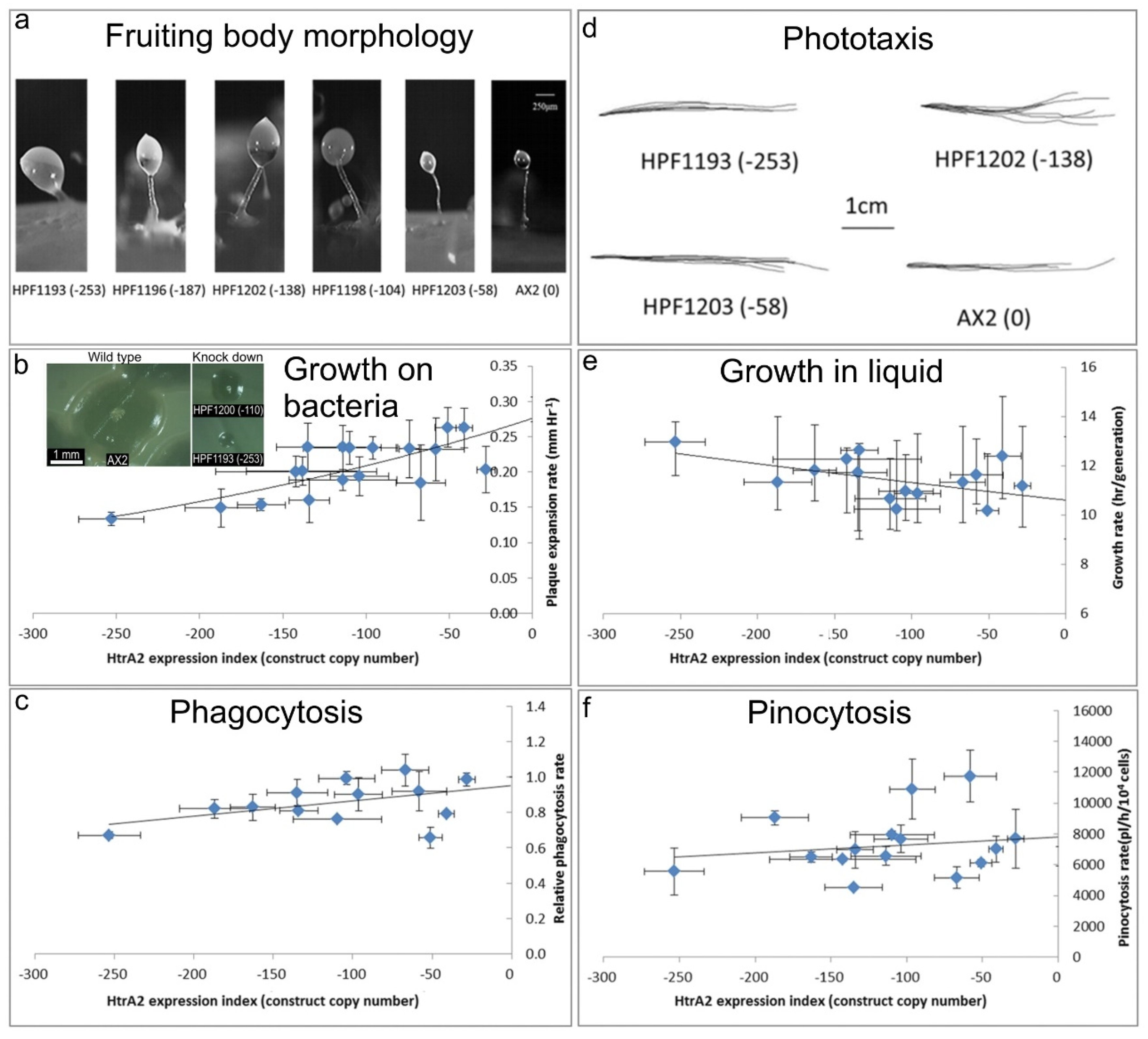
Figure 4).
(a) Aberrant fruiting body morphology.
Wild type AX2 fruiting bodies have long and thin stalks, whereas the antisense transformants produced larger fruiting bodies with short, thick stalks. The severity of the defect correlates with decreasing HTRA2 expression level as indicated by the copy numbers of the HTRA2 antisense-inhibition construct (pPROF689—copy numbers in parentheses).
(b) Slower rate of plaque expansion on bacterial lawns.
Plaque sizes were measured over several days, the plaque expansion rates calculated by linear regression and then plotted against the expression index for HTRA2 (pPROF689 copy numbers). The plaque expansion rate of wild type AX2 was less than 0.3 mm hr−1. The plaque expansion rates decreased as the level of HTRA2 expression decreased. The quadratic regression was significant at p = 2.22 × 10−4 (F test, n = 19). Error bars are standard errors of the mean from four independent experiments. The inset shows as examples, single colonies of AX2 and two knockdown strains after five days incubation on streak dilution plates on lawns of E. aerogenes (construct copy numbers in parentheses).
(c) Normal rate of phagocytosis.
The phagocytosis rates of HTRA2 knockdown strains were measured as the rate of uptake of fluorescent E. coli DsRed cells and normalized against AX2. There was no significant correlation of phagocytosis rates with the HTRA2 expression level (p = 0.076, F test, n = 14). Error bars are standard errors of the mean from three independent experiments.
(d) Normal phototaxis by multicellular slugs.
The slug trails of parental strain AX2 and the HTRA2 antisense transformants containing different copy numbers of pPROF689 as indicated in brackets. The light source was located to the right of the figure.
(e) Slightly slower growth in liquid.
The knockdown strains grew at a slightly slower rate than AX2 (p = 0.0193, F test, n = 16). Error bars are standard errors of the mean from three independent experiments.
(f) Normal rate of pinocytosis.
The knockdown strains showed no significant reduction in pinocytosis rates (rate of uptake of FITC-containing medium) as the severity of the antisense inhibition increased (p = 0.5597, F test, n = 14). Error bars are standard errors of the mean from three independent experiments.
3.3.1. Fruiting Body Morphology Is Regulated by HTRA2
Mitochondrial dysfunction in
D. discoideum results in defective fruiting body morphology– culminants have shorter and thicker stalks and this phenotype is mediated by chronic activation of AMPK [
20,
41,
42]. This phenotype was examined in the HTRA2 antisense-inhibited transformants to determine if it would phenocopy mitochondrial dysfunction. The morphology of AX2 and HTRA2 antisense-inhibited transformants was examined after growth and development on bacterial lawns on nutrient agar (SM) plates. (
Figure 4a) shows that the reduction in HTRA2 expression resulted in aberrant fruiting bodies with shorter and thicker stalks compared to AX2. The severity of this abnormal morphology was correlated with the copy numbers of the HTRA2 antisense-inhibition construct (pPROF689). It resembles the morphology of mitochondrially diseased
D. discoideum strains, a result consistent with the possibility that HTRA2 loss impairs mitochondrial respiratory function.
3.3.2. Knocking Down HTRA2 Expression Inhibits Plaque Expansion in Dictyostelium discoideum but Does Not Affect Phagocytosis Rates
Another defective phenotype that characterizes mitochondrial disease in
Dictyostelium is impaired plaque expansion or growth on
E. coli B2 lawns. HTRA2 antisense-inhibited strains displayed decreased plaque expansion rates that correlated with the construct copy number (
Figure 4b). At the highest copy numbers of the antisense construct, the plaque expansion rate was approximately halved. This reduced rate of growth on bacterial lawns could not be explained by a defect in phagocytosis (
Figure 4c).
3.3.3. Axenic Growth Is Affected Only Slightly and Pinocytosis Is Unaffected by HTRA2 Expression Levels
Laboratory strains of
D. discoideum are able to grow on solid media and also axenically in liquid media. To determine if HTRA2 plays a role in growth in liquid media (axenic growth), AX2 and the knockdown strains were inoculated into sterilized HL-5 and the generation time was measured. The results showed that the generation times of HTRA2 antisense transformants are affected only slightly by reduced expression of HTRA2, with generation times being only about 20% longer even at the highest copy numbers of the antisense construct (
Figure 4e). Growth in liquid medium by
Dictyostelium cells is dependent on nutrient uptake by pinocytosis and when this was measured we found no significant effect of HTRA2 expression on the rates of pinocytosis (
Figure 4f). This differs from the phenotype displayed by mitochondrially diseased
D. discoideum strains whose growth typically is dramatically slower in liquid medium, although unaccompanied by corresponding defects in pinocytosis [
20].
3.3.4. HTRA2 Expression Levels Have No Effect on Phototaxis
The photosensory signalling pathway in
D. discoideum is sensitive to mitochondrial respiratory dysfunction so that, with one exception, all tested genetic causes of mitochondrial dysfunction result in impaired orientation during slug phototaxis towards a lateral light source [
20]. HTRA2 is a nuclear-encoded mitochondrial protein, and its inhibition may affect mitochondrial function. Therefore, we tested the phototactic ability of the HTRA2 antisense-inhibited transformants and found that the antisense transformants showed accuracies of phototaxis resembling the wild type AX2 (
Figure 4d). This result is not consistent with HTRA2 knockdown causing impairment of mitochondrial respiratory function.
3.4. Mitochondrial Respiration Is Not Significantly Affected by Knockdown of HTRA2
The foregoing results showed that reduced levels of HTRA2 expression cause a pattern of phenotypes that exhibits only some of the defects typically observed in mitochondrially diseased
Dictyostelium strains, namely aberrant morphogenesis and reduced growth rates, unaccompanied by defects in endocytosis. However, there is no defect in phototaxis and the defect in growth in liquid medium is only mild. These differences suggest that although HTRA2’s role in the mitochondria is positive and protective, its loss evokes cytopathological mechanisms that differ from the respiratory defects typical of mitochondrial disease. To verify that HTRA2 loss does not cause mitochondrial respiratory defects, we used Seahorse respirometry as previously described [
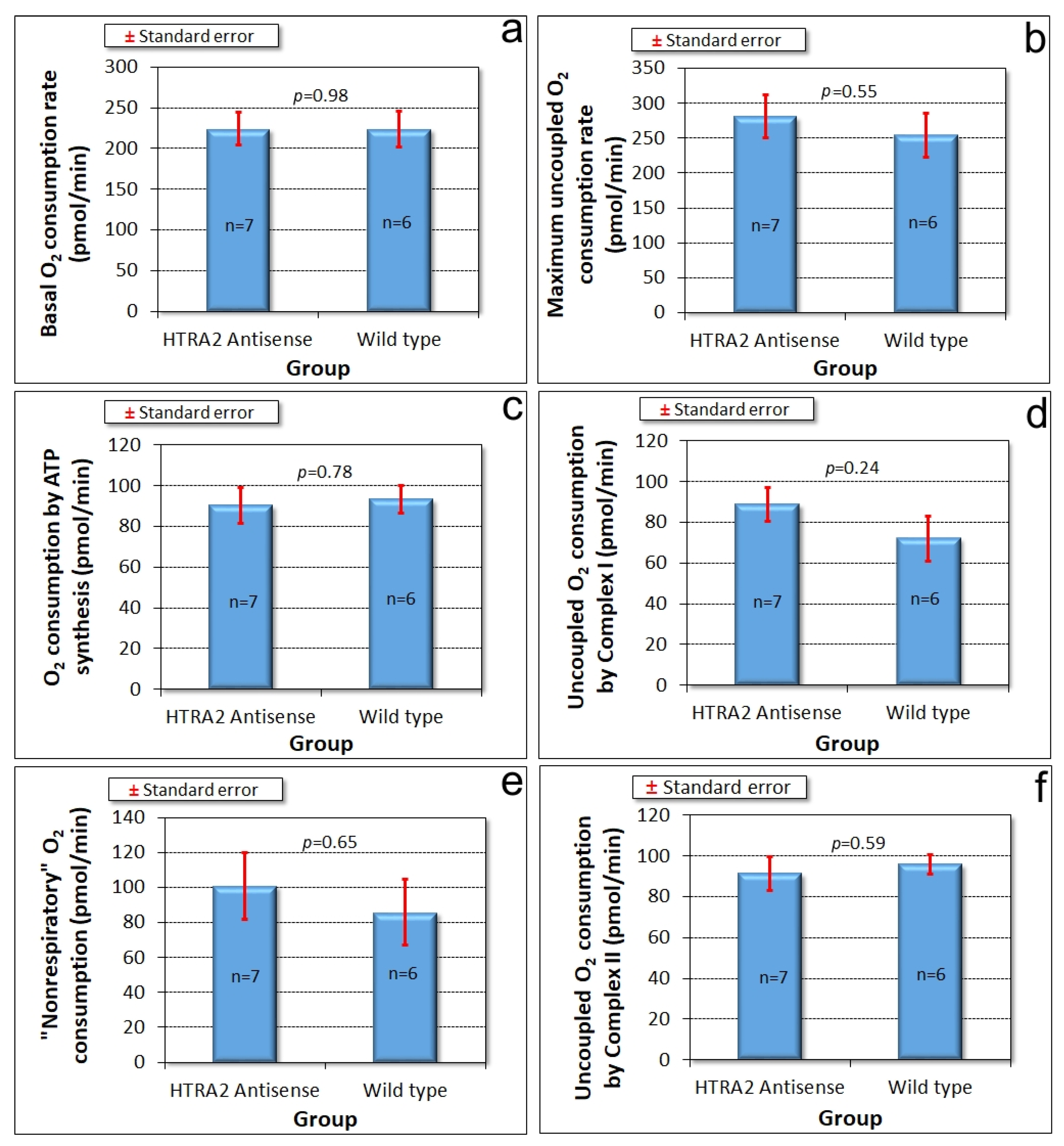
31] to measure mitochondrial respiratory activity directly. The results showed that HTRA2 knockdown does not cause any significant loss of mitochondrial respiratory function (
Figure 5).
3.5. Overexpression of HTRA2 Is Toxic to Dictyostelium discoideum Cells and This Toxicity Depends on Its Serine Protease Activity
3.5.1. Dictyostelium Transformants Overexpressing Wild Type HTRA2 Cannot Be Isolated
To investigate the positive, protective role of HTRA2 further, we attempted to ectopically overexpress functional wild type HTRA2 in
Dictyostelium cells. We created an HTRA2 overexpression construct (pPROF691) for expression of the full length wild type
htrA gene under the control of the actin-15 promoter (A-15P). This was used to transform the parental strain AX2. These transformants were very difficult to isolate, averaging less than 10 per experiment, compared to hundreds in a single more typical transformation. After multiple experiments, 50 transformants were isolated and purified. PCR assays showed that only about half (26) of these contained the full length recombinant
htrA gene. In the remainder, the recombinant gene was either undetectable by PCR or contained large deletions (
Text S1, Figure S3). Of those transformants that contained the full length recombinant
htrA, half had unusually low numbers of copies of the
htrA amplicon, not very different from the single endogeneous copy of the gene (
Text S1–3, Table S2). Three of the ca. 25% of transformants with moderate copy numbers of the the full length recombinant gene were studied further and shown to contain mutations in
htrA (single base substitutions or insertions causing reading frame shifts and truncation of the protein) (
Text S3, Figure S4). In combination with the large deletions demonstrated in most transformants, and the low copy numbers in most of the remainder, the mutations in these sequenced cases suggest that the serine protease activity of HTRA2 may be lethal in overexpression transformants because of uncontrolled proteolysis. The only survivors were the small fraction of transformed cells in which the
htrA gene was poorly expressed or had been mutated or deleted.
3.5.2. Dictyostelium discoideum HTRA2: GFP Fusion Transformants Do Not Express Functional GFP Fusion Protein
Not only were we unable to obtain transformants ectopically overexpressing the wild type HTRA2, we also were unable to isolate overexpressers of the wild type protein tagged at the C-terminus with GFP. Our intent had been to verify the subcellular localization of HTRA2. For this purpose, a construct expressing GFP-tagged HTRA2 (pPROF694) was transformed into wild type
D. discoideum AX2 and analysed with fluorescent microscopy. We did obtain transformants, but there was no GFP fluorescence in any of them (e.g.,
Figure S5).
To determine why the transformants displayed no fluorescence, the presence of the GFP and
htrA sequences were verified by PCR. The results showed that both the
htrA and the GFP portions of the HTRA2:GFP fusion gene in the construct (pPROF694) were present in their entirety in the transformants (
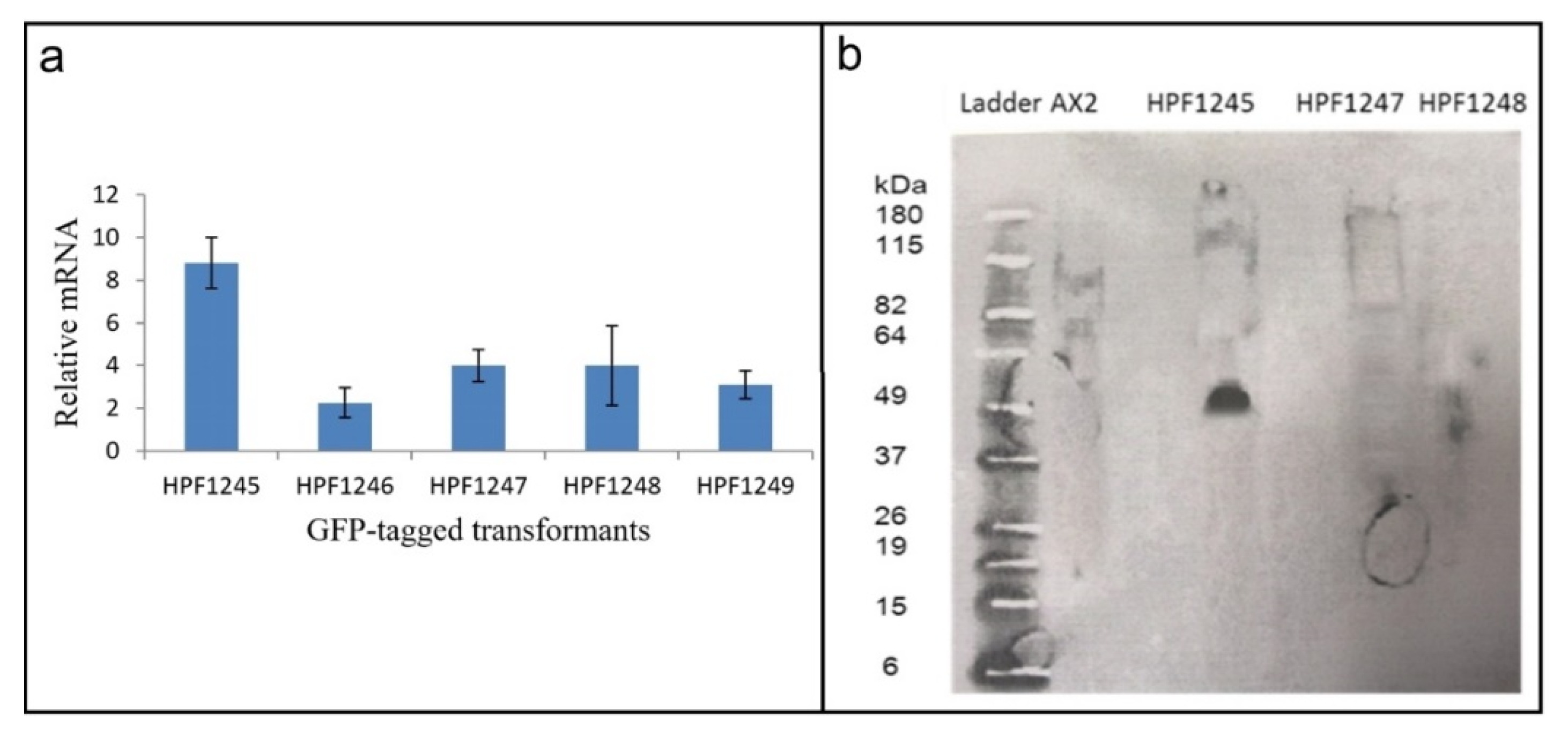
Figure S6). We therefore determined if the GFP sequence in these transformants was expressed at both the RNA (qRT-PCR) (
Figure 6a) and protein (Western blot) (
Figure 6b) level.
RNA was extracted from three HTRA2:GFP fusion transformants and from two DJ-1: GFP transformants [
28] which were to be used as positive controls (as these do display GFP fluorescence). qRT-PCR was carried out to check expression of the GFP portion of the fusion mRNA using filamin as a reference gene [
45]. The result showed that the recombinant GFP fusion gene was transcribed into mRNA (
Figure 6a). To determine if the mRNA was translated, a Western blot was performed using an anti-GFP antibody. The blot showed that the GFP protein was not expressed in the HTRA2:GFP transformants, but was expressed in the DJ-1:GFP transformant [
28] used as a positive control (
Figure 6b). In view of the apparent toxicity of the wild type HTRA2, these results suggest that the GFP portion of the fusion protein was not expressed because of mutations and premature translational stops in the upstream HTRA2 portion of the protein.
3.6. A Protease-Dead Mutant (S300A) of Dictyostelium HTRA2 Can Be Expressed in Dictyostelium discoideum
The foregoing results suggested strongly that overexpression of active HTRA2 is toxic to
D. discoideum cells, perhaps due to the uncontrolled serine protease activity. To confirm if this is true, an essential serine residue in the catalytic site of
D. discoideum HTRA2 was selected and mutated to ablate its serine protease activity. Suzuki et al. [
35] showed that human HTRA2 contains two main domains: the N-terminal serine protease domain which prevents the accumulation of misfolded proteins and the C-terminal PDZ domain which modulates the protease activity. According to Polgár [
46], the trypsin-like proteases contain a catalytic triad of serine, histidine and aspartic acid residues that exhibit similar spatial arrangements in the active site. The serine protease domain of HTRA2 adopts the same fold as trypsin including the essential, highly conserved, catalytic serine (S306 in humans, S300 in
D. discoideum) (
Figure 1) [
47,
48]. Others have shown that S306 in the mammalian protein is essential for the proteolytic and protective activities of HTRA2. Therefore, we created a protease-dead mutant of the
Dictyostelium HTRA2.
To remove the serine protease activity of
D. discoideum HTRA2, serine 300 (
Figure 1) was replaced by alanine and the construct pPROF692 expressing the mutant form of the protein, HTRA2
S300A, was transformed into the wild type
D. discoideum AX2. A total of 280 transformants were obtained from a single (typical) transformation. To determine if the full HTRA2
S300A sequence was present in these transformants, the genomic DNA (gDNA) of 50 randomly chosen transformants was extracted and used as the template to amplify the full length recombinant gene. The results showed that all 50 tested transformants contained full length gene encoding HTRA2
S300A, in stark contrast to the earlier results with the wild type HTRA2. Quantitative PCR showed that the copy numbers ranged from 42 to 312, a much more typical range for this kind of experiment. The expression of HTRA2
S300A mRNA was also measured using qRT-PCR and was strongly correlated with the construct copy number as expected (
Figure S7). Taking all of the results for overexpression of the wild type and mutant forms of HTRA2 together, we conclude that wild type HTRA2 cannot be overexpressed in
Dictyostelium due to the toxicity caused by high levels of protease activity.
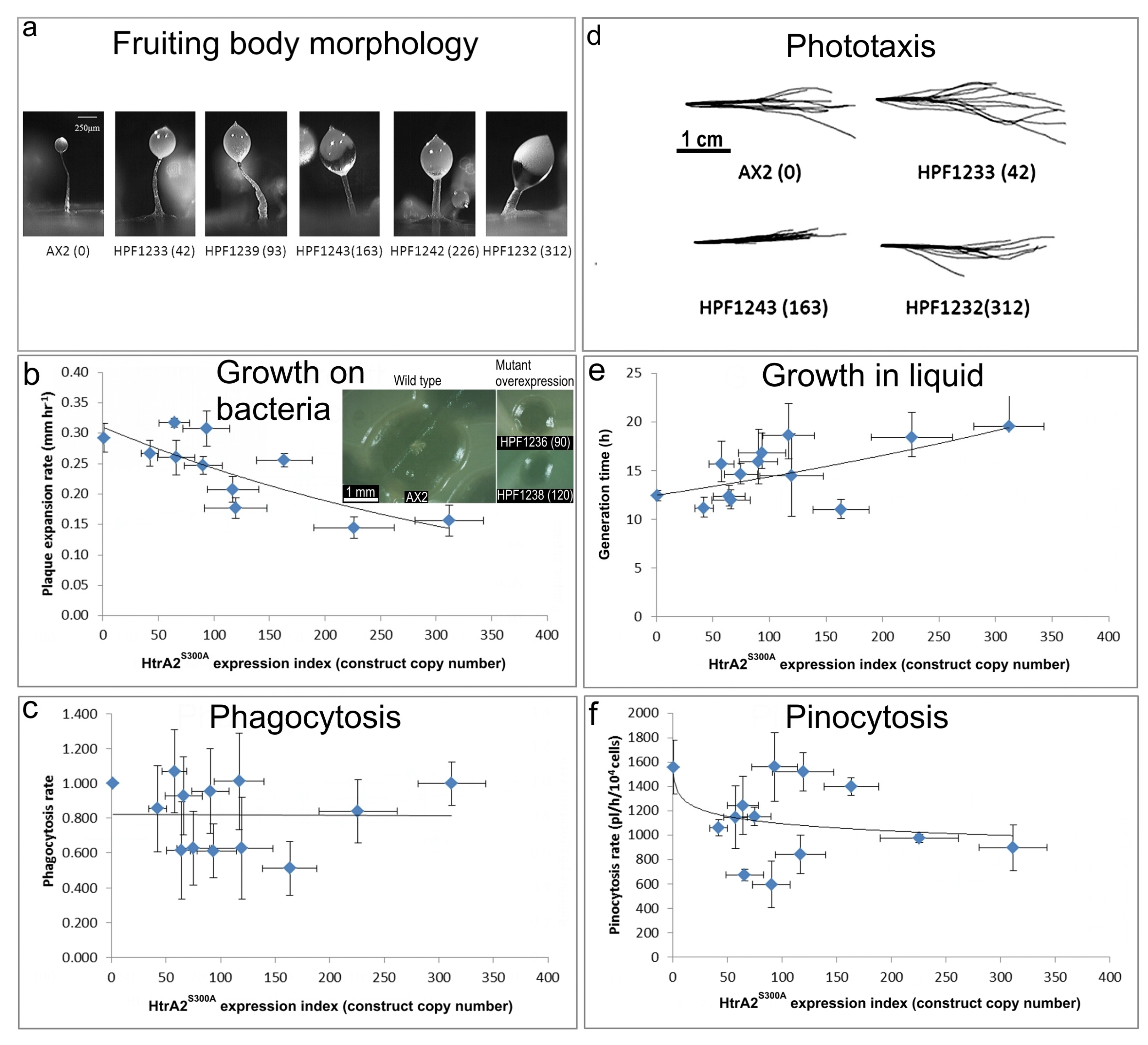
3.7. Phenotypic Analysis of HTRA2S300A Transformants
The inability to overexpress HTRA2 in
D. discoideum was due to the activity of the serine protease domain. By mutating the essential catalytic serine S
300 in this domain, we were able to obtain overexpression transformants. When we fused this mutant form of the protein to GFP, we were also able to obtain recombinant GFP-expressing forms that confirmed the mitochondrial localization of the protein (
Figure 2). Since HTRA2 is reported to exert a protective effect in the mitochondria, it was possible that overexpression of HTRA2
S300A would enhance this protective role. This would indicate that the protective functions of the protein in the mitochondria are not dependent on the protease activity. However, others have reported that HTRA2’s protective functions are lost when the protease activity is ablated.
HTRA2 is a homotrimeric protease whose three PDZ protein-binding domains recruit a limited range of target substates and, upon binding them, activate the catalytic protease domain. Ectopic overexpression of a protease-dead form of the protein could result in competitive inhibition of the native wild type protein that is still expressed from the endogeneous
htrA gene. This could be a result of competition for substrates and/or formation of inactive mutant/wild type heterotrimers. In either case, the overexpression of the protease-dead protein should phenocopy knockdown of expression of the endogeneous gene. To investigate whether this is so, we determined the phenotypes of transformants overexpressing HTRA2
S300A (
Figure 7).
(a) Aberrant fruiting body morphology.
The stalks of HTRA2S300A transformant fruiting bodies were thick and short compared to the thin and long stalk of the AX2 fruiting body. This phenotype became more severe with an increase of HTRA2S300A expression levels indicated by increasing copy numbers of pPROF692 in parentheses after the strain names of the individual transformants.
(b) Slower rate of plaque expansion on bacterial lawns.
The plaque expansion rate of AX2 was 0.29 mm hr−1. Compared to AX2, 80% of transformants containing mutated HTRA2 displayed slower plaque expansion rates and the plaque expansion rates reduced with increasing HTRA2S300A expression levels (regression of plaque expansion rate against pPROF692 (HTRA2S300A) copy numbers highly significant: p = 0.00396, F test, n = 11). Error bars are standard errors of the mean from four replicate measurements. The inset shows as examples, single colonies of AX2 and two HTRA2S300A overexpression strains after five days incubation on streak dilution plates on lawns of E. aerogenes (construct copy numbers in parentheses).
(c) Normal rate of phagocytosis.
The phagocytosis rates of the transformants were not significantly affected by the HTRA2S300A expression levels (regression not significant: p = 0.982, F test, n = 13). Error bars are standard errors of the mean from three independent experiments.
(d) Normal phototaxis by multicellular slugs.
Slugs of AX2 and transformants containing different numbers of copies of the HTRA2S300A expression construct (shown in parentheses) migrated to the light source which was to the right of the figures. Like the parental AX2 slugs, the slugs of the transformants displayed normal phototaxis.
(e) Slightly slower growth in liquid.
The average generation time of HTRA2S300A transformants was 15.6 + 0.9 h, significantly longer than the generation time of AX2 (10.4 + 2.6 h). The generation times of HTRA2S300A transformants were weakly correlated with mHTRA2 expression levels as indicated by the copy numbers of pPROF692 expressing HTRA2S300A and the correlation was statistically significant (Pearson correlation coefficient ρ = 0.58, p = 0.03, n = 11). Error bars are standard errors of the mean from three independent experiments.
(f) Normal rate of pinocytosis.
The pinocytosis rates of HTRA2S300A overexpression transformants decreased slightly with increased mutant HTRA2 expression levels as indicated by increasing pPROF692 copy numbers, but the regression was not significant compared to wild type AX2 (Pearson correlation coefficient ρ = 0.23, p = 0.22, n = 13; log-linear regression not significant at p = 0.209, F test, n = 13). Error bars are standard errors of the mean from three independent experiments.
3.7.1. Overexpression of Protease-Dead HTRA2 Resulted in Defective Fruiting Body Morphology
Knockdown of endogeneous HTRA2 expression caused the formation of abnormal fruiting bodies. To determine if morphogenesis was affected similarly by overexpression of HTRA2
S300A, the fruiting body morphology of the HTRA2 transformants was examined. The mutant overexpression strains had altered morphologies with larger sori and shorter, thicker stalks. The severity of this defect correlated with the HTRA2
S300A expression index (
Figure 7a). This is similar to the defects resulting from HTRA2 knockdown and reminiscent of what occurs in mitochondrially diseased
Dictyostelium strains, supporting the possibility that overexpression of mutant HTRA2 may result in altered mitochondrial function.
3.7.2. Plaque Expansion Was Inhibited but Phagocytosis Was Unaffected by Overexpression of HTRA2S300A
The growth of HTRA2
S300A expression strains on
E. coli B2 lawn was measured and the results show that the plaque expansion rates decreased with increased expression levels of protease-dead HTRA2 (
Figure 7b). To determine if this was due to a decreased ability to take up nutrients, phagocytosis rates were measured and plotted against the expression index (copy numbers) of HTRA2
S300A. The results show that phagocytosis rates were unaffected in the HTRA2
S300A transformants (
Figure 7c). This implies that the growth defect is not due to impaired phagocytosis, as was also the case for the HTRA2 knockdown strains.
3.7.3. Growth in Liquid Is Slower While Pinocytosis Is Not Significantly Inhibited by Overexpression of HTRA2S300A
To determine if overexpression of protease-dead HTRA2 also affected axenic growth, the growth rates of the HTRA2
S300A transformants were measured in liquid media. Their generation times were calculated and plotted against the HTRA2
S300A expression index. The generation time of HTRA2
S300A transformants did increase and the severity of this defect was correlated with the HTRA2 expression index (
Figure 7e).
To determine if there was a defect in the transformants’ ability to take up liquid nutrients, the pinocytosis rates were measured. The (
Figure 7f) shows that although the HTRA2
S300A transformants appeared to exhibit slightly slower pinocytosis rates, the effect was not statistically significant.
3.7.4. Phototaxis and Mitochondrial Respiration Are Unaffected by Overexpression of Protease-Dead HTRA2S300A
Amoebae of AX2 and HTRA2
S300A overexpression transformants were inoculated onto charcoal agar plates without any food supply, allowed to form slugs and migrate towards a lateral light source. The slug trails were then blotted onto PVC (Polyvinyl chloride) discs, stained with Coomassie Brilliant Blue R and their trails were digitized. (
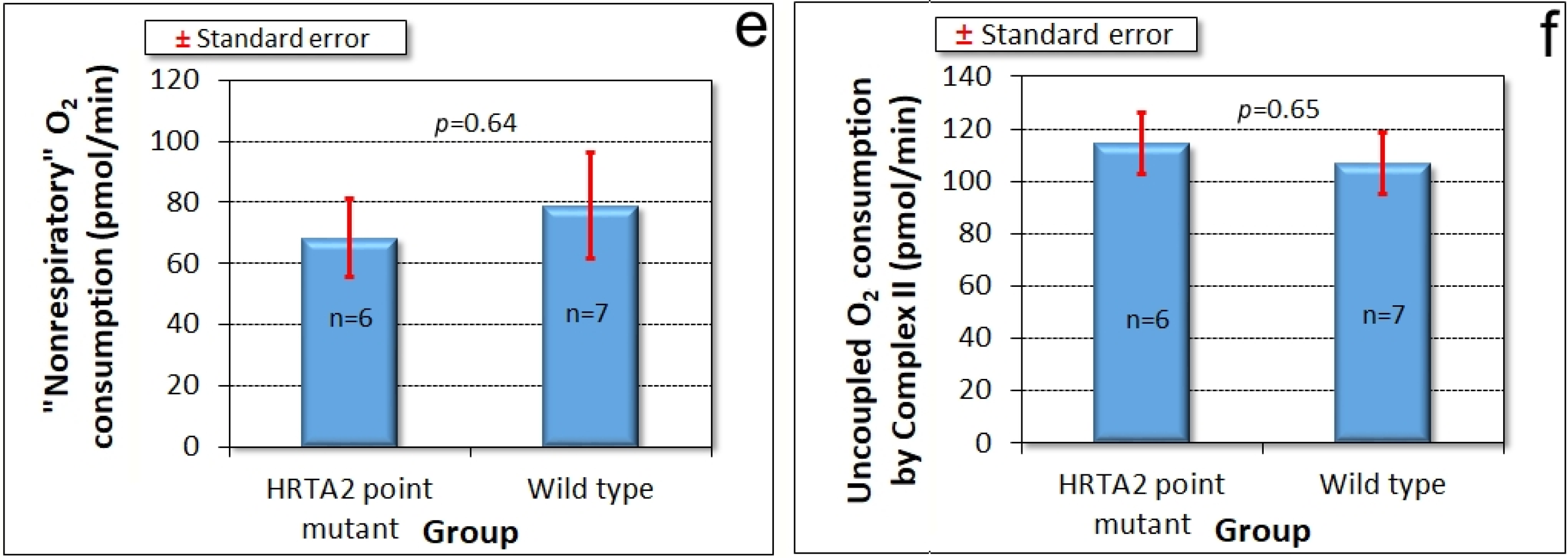
Figure 7d) shows that phototaxis was not affected by overexpression of the mutant protease. As with the knockdown strains, the absence of a phototaxis defect is not consistent with an impairment of mitochondrial respiration and the absence of a mitochondrial respiratory defect was directly confirmed using Seahorse respirometry (
Figure 8).
4. Discussion
The results reported here suggest that
Dictyostelium HTRA2 is located in the mitochondria, from which location it plays a positive, cytoprotective role. We were able to demonstrate experimentally the subcellular location only of the GFP-tagged, overexpressed HTRA2
S300A. However, there is no reason to expect that the wild type protein would localize differently, especially since it contains a recognizable N-terminal mitochondrial leader peptide and its human homologue has been reported to be mitochondrial [
37]. This being so, cytopathology caused by genetic loss or reduction of HTRA2 activity constitutes a mitochondrial disease and as such has provided some of the evidence that Parkinson’s disease involves mitochondrial defects. However, the effects of HTRA2 loss on mitochondrial function remain unclear. For example, Yun et al. [
19] suggested that HTRA2 did not act downstream of PINK1 in
Drosophila as HTRA2 knockout mutants did not display the defective mitochondrial integrity and dynamics observed in PINK1 null mutants. Uncertainties regarding the cytopathological roles of HTRA2 mutations may result from the complexities of mitochondrial disease. Alternatively, they may be due to different experimental conditions (such as whether or not oxidative stress was applied) or different phenotypes being analyzed (such as mitochondrial integrity and dynamics, degeneration in direct flight muscles, mitochondrial morphology, male sterility and so on). To investigate the cytopathological roles of mitochondrial HTRA2, a model providing consistent phenotype-genotype correlations in mitochondrial dysfunction would be an advantage.
The
Dictyostelium model provides such an advantage, so in this work we asked whether the cytopathological outcomes of HTRA2 loss in
Dictyostelium are consistent with and caused by impaired mitochondrial respiration. We found that genetically impaired HTRA2 function (by antisense inhibition or presumed competitive inhibition by an ectopically overexpressed mutant form) had no effect on phototaxis, but caused defects in morphogenesis and growth (particularly on bacterial lawns), unaccompanied by corresponding defects in endocytosis. The slow plaque expansion rates on bacterial lawns could be due to slow growth or impaired amoeboid motility or both. This pattern of phenotypes is only partly reminiscent of the typical outcomes of mitochondrial respiratory disease in
Dictyostelium, which are caused by chronic hyperactivity of the energy stress-sensing protein kinase AMPK [
20,
41,
42,
43,
44]. This produces a variety of AMPK-mediated phenotypes, including impaired phototaxis, morphogenesis and equally severe defects in growth both on bacteria and in liquid medium [
20]. The growth defects are not caused or accompanied by corresponding defects in endocytosis [
20].
Since HTRA2 knockdown does not faithfully phenocopy mitochondrial respiratory disease, the defects it causes seemed unlikely to result from impaired mitochondrial respiration. Direct measurement of mitochondrial respiratory activity confirmed that mitochondrial respiration was not inhibited either by HTRA2 knockdown, or by overexpression of the inhibitory, protease-dead HTRA2S300A. Despite its mitochondrial location, HTRA2 is thus not essential for normal mitochondrial respiratory activity, suggesting that its protective role may instead be directed primarily towards other mitochondrial functions. The possibilities could include lipid, amino acid or iron metabolism as well as signalling by small molecules such as Ca2+, succinate or reactive oxygen species (ROS). In all of these cases, the resulting dysregulation could in turn account (indirectly) for the impaired growth that was observed in the antisense transformants.
HTRA2 loss also partly phenocopies the loss of another PD-associated protein, DJ-1, which also has no effect on phototaxis, but severely impairs growth on bacteria and only slightly impairs growth in liquid [
28]. In the case of DJ-1 loss, however, the growth defects are accompanied and presumably caused by a corresponding deficiency in the requisite endocytic pathway. These results suggest distinct, but intersecting pathways underlying the cytopathology of different forms of mitochondrial disease and of PD. It will be of interest in the future to determine if chronic AMPK hyperactivity contributes to the aberrant phenotypes in cells with reduced HTRA2 activities.
Despite its positive protective role in the mitochondria, we found that wild type HTRA2 is cytotoxic when ectopically overexpressed. This was revealed by the difficulty of obtaining HTRA2 overexpression transformants, by the unusually low copy numbers of most transformants that we were able to obtain, by the mutations in the HTRA2 coding sequence in those few transformants with higher copy numbers and by the absence of expression and fluorescence of GFP in those HTRA2:GFP transformants that could be isolated. Mutation of the serine protease domain overcame this, so that many transformants expressing HTRA2S300A were obtained and several were further studied.
It has been suggested that, as a serine protease, HTRA2 plays its crucial protective role by contributing to protein quality control in the mitochondria [
12,
49,
50]. This protective proteolytic function might also apply to
D. discoideum and, as has been suggested for other organisms, could explain why HTRA2 loss causes phenotypic defects. On the other hand, when HTRA2 was overexpressed, the excess HTRA2 may have degraded essential proteins in the mitochondria thereby initiating cell death. Alternatively, HTRA2 overexpression might have caused an imbalance in HTRA2 levels inside and outside the mitochondria. Although HTRA2 usually localizes to the mitochondria, its overexpression could have resulted in incomplete import of the protein into the mitochondria or its partial release from the mitochondria to the cytosol where it may have initiated cell death. However, this possibility is not supported by the fact that overexpressing the GFP-tagged, protease-dead HTRA2
S300A did not reveal any significant failure of targeting of the overexpressed protein to the mitochondria.
The mitochondrial localization of HTRA2
S300A, even when overexpressed, is consistent with findings in other organisms and confirms the in silico predictions of a mitochondrial location for this protein. Thus, the serine protease activity of HTRA2 exerts a double-edged function in the mitochondria—it usually protects the cells, presumably by removing denatured mitochondrial proteins, but its hyperactivity in the mitochondria is also cytotoxic. One implication is that, while loss of function mutations in HTRA2 can cause cellular pathology, mutations that upregulate its protease activity could also do so, albeit by a different mechanism. For example, a P143A substitution results in hyperphosphorylation (and presumably elevated activity) as well as increased neurotoxicity of human HTRA2 [
27]. This has been suggested to have contributed to PD pathology and mitochondrial dysfunction in a Taiwanese patient carrying this allele [
27]. The PDZ domain of HTRA2 is required for keeping the protease activity of the protein in check and for recruitment of the correct substrates [
48]. Its loss could produce less discriminating, elevated proteolytic activity that would also cause mitochondrial dysfunction. Both of the phosphorylatable serines in HTRA2 that have been implicated in PD (S142 and S400) are conserved in
Dicyostelium (
Figure 1). It would be valuable in future work to study their roles in this model.
In mammalian cells, HTRA2 has been reported to translocate from the mitochondria to the cytosol under stress conditions, where it initiates both caspase-dependent and -independent cell death pathways [
2]. However, our results suggest that overactive HTRA2 is lethal to cells even in its original mitochondrial location. This is consistent with the recent report that overexpressed, wild type HTRA2 is targeted to the mitochondria, causing a motor defect and cell death in the brains of transgenic mice [
51]. Like us, these authors also found that a PD-associated point mutant form of HTRA2 (HTRA2
G399S) was targeted to the mitochondria and caused a dominant loss of function when overexpressed. It will be of interest in future work to determine if
Dictyostelium HTRA2 also relocates to the cytosol under stress conditions and whether that has different cytopathological consequences.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








