Assembly of the Mitochondrial Genome in the Campanulaceae Family Using Illumina Low-Coverage Sequencing

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Whole-Genome Sequencing
2.2. Assembly of Mitochondrial Genomes
2.3. Validation of Mitochondrial Genomes
2.4. Annotation of Mitochondrial Genome
2.5. Analysis of Repetitive Sequences
2.6. Bayesian Phylogenetic Analysis
3. Results
3.1. Pipeline for the Assembly
3.2. Complete Mitochondrial Genomes
3.3. Annotated Genes in the Mitochondrial Genomes
3.4. Repetitive Sequences in the Mitochondrial Genomes
3.5. Phylogenetic Relationships Based on Mitochondrial Sequences
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lammers, T.G. The Families and Genera of Vascular Plants; Kadereit, J.W., Bittrich, V., Eds.; Springer: New York, NY, USA, 2007; Volume 8, pp. 26–56. [Google Scholar]
- Hong, D.; Song, G.; Lammers, T.G.; Klein, L. Flora of China; Zhengyi, W., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden: St. Louis, MO, USA, 2011; Volume 19. [Google Scholar]
- Halevy, A.H.; Shlomo, E.; Ziv, O. Improving cut flower production of balloon flower. HortScience 2002, 37, 759–761. [Google Scholar]
- Zhang, L.; Wang, Y.; Yang, D.; Zhang, C.; Zhang, N.; Li, M.; Liu, Y. Platycodon grandiflorus—An ethnopharmacological, phytochemical and pharmacological review. J. Ethnopharmacol. 2015, 164, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Hossen, M.J.; Kim, M.Y.; Kim, J.H.; Cho, J.Y. Codonopsis lanceolata: A review of its therapeutic potentials. Phytother. Res. 2016, 30, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, B.B.; Gruissem, W.; Jones, R.L. Biochemistry & Moleculashsar Biology of Plants; Jones, R.L., Buchanan, B.B., Gruissem, W., Eds.; American Society of Plant Physiologists: Rockville, MD, USA, 2000; Volume 40. [Google Scholar]
- Gualberto, J.M.; Newton, K.J. Plant mitochondrial genomes: Dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef] [PubMed]
- Iorizzo, M.; Senalik, D.; Szklarczyk, M.; Grzebelus, D.; Spooner, D.; Simon, P. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 2012, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yang, A.; Lv, J.; Gong, D.; Sun, Y. The complete mitochondrial genome sequence of Sua-type cytoplasmic male sterility of tobacco (Nicotiana tabacum). Mitochondrial DNA Part A 2016, 27, 2929–2930. [Google Scholar] [CrossRef] [PubMed]
- Hajibabaei, M.; Singer, G.A.; Hebert, P.D.; Hickey, D.A. DNA barcoding: How it complements taxonomy, molecular phylogenetics and population genetics. TRENDS Genet. 2007, 23, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.; Smit, A.F.; Hood, L.; Glusman, G. Realistic artificial DNA sequences as negative controls for computational genomics. Nucleic Acids Res. 2014, 42, e99. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.; Flores-Vergara, M.; Krasynanski, S.; Kumar, S.; Thompson, W. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.-C.; Lee, J.; Yu, Y.; Yang, K.; Choi, B.-S.; Koh, H.-J.; Waminal, N.E.; Choi, H.-I.; Kim, N.-H. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza Aa genome species. Sci. Rep. 2015, 5, 15655. [Google Scholar] [CrossRef] [PubMed]
- Myers, E.W.; Sutton, G.G.; Delcher, A.L.; Dew, I.M.; Fasulo, D.P.; Flanigan, M.J.; Kravitz, S.A.; Mobarry, C.M.; Reinert, K.H.; Remington, K.A. A whole-genome assembly of Drosophila. Science 2000, 287, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. Soapdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Jayakumar, V.; Nitasaka, E.; Toyoda, A.; Noguchi, H.; Itoh, T.; Shin, T.; Minakuchi, Y.; Koda, Y.; Nagano, A.J. Genome sequence and analysis of the Japanese morning glory Ipomoea Nil. Nat. Commun. 2016, 7, 13295. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. 2003, 10.3.11–10.3.18. [Google Scholar] [CrossRef] [PubMed]
- Grassa, C.J.; Ebert, D.P.; Kane, N.C.; Rieseberg, L.H. Complete mitochondrial genome sequence of sunflower (Helianthus Annuus L.). Genome Announc. 2016, 4, e00981-16. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, D.; Schlautman, B.; Steffan, S.; Polashock, J.; Vorsa, N.; Zalapa, J. The american cranberry mitochondrial genome reveals the presence of selenocysteine (tRNA-Sec and SECIS) insertion machinery in land plants. Gene 2014, 536, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. Organellargenomedraw (Ogdraw): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. Beast 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.-F.; Kerrest, A.; Dujon, B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol. Mol. Biol. Rev. 2008, 72, 686–727. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.-C.; Lee, J.; Lee, H.O.; Joh, H.J.; Kim, N.-H.; Park, H.-S.; Yang, T.-J. Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax Ginseng species. PLoS ONE 2015, 10, e0117159. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-P.; Lo, H.-F.; Chen, C.-Y.; Chen, L.-F.O. The complete mitochondrial genome of mungbean Vigna radiata Var. radiata NM92 and a phylogenetic analysis of crops in angiosperms. Mitochondrial DNA Part A 2016, 27, 3731–3732. [Google Scholar]
- Petersen, G.; Cuenca, A.; Zervas, A.; Ross, G.T.; Graham, S.W.; Barrett, C.F.; Davis, J.I.; Seberg, O. Mitochondrial genome evolution in Alismatales: Size reduction and extensive loss of ribosomal protein genes. PLoS ONE 2017, 12, e0177606. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, F.; Hu, K. The complete mitochondrial genome sequence of an annual wild tobacco Nicotiana attenuata. Mitochondrial DNA Part B 2017, 2, 924–925. [Google Scholar] [CrossRef]
- Ye, N.; Wang, X.; Li, J.; Bi, C.; Xu, Y.; Wu, D.; Ye, Q. Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. PeerJ 2017, 5, e3148. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.R.; Alvarenga, D.O.; Aranguren, Y.; Penha, H.A.; Fernandes, C.C.; Pinheiro, D.G.; Oliveira, M.T.; Michael, T.P.; Miranda, V.F.; Varani, A.M. The mitochondrial genome of the terrestrial carnivorous plant Utricularia reniformis (Lentibulariaceae): Structure, comparative analysis and evolutionary landmarks. PLoS ONE 2017, 12, e0180484. [Google Scholar] [CrossRef] [PubMed]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Rosenblueth, M.; Qiu, Y.-L.; Palmer, J.D. Multiple losses and transfers to the nucleus of two mitochondrial succinate dehydrogenase genes during angiosperm evolution. Genetics 2001, 158, 1289–1300. [Google Scholar] [PubMed]
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes—Though this be madness, yet there’s method in’t. J. Exp. Bot. 2010, 61, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2012, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.-D.; Shao, R.; Yuan, M.-L.; Dou, W.; Barker, S.C.; Wang, J.-J. The multipartite mitochondrial genome of Liposcelis bostrychophila: Insights into the evolution of mitochondrial genomes in bilateral animals. PLoS ONE 2012, 7, e33973. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, Y.; Watase, Y.; Nagase, M.; Makita, N.; Yagura, S.; Hirai, A.; Sugiura, M. The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: Comparative analysis of mitochondrial genomes in higher plants. Mol. Genet. Genom. 2005, 272, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Ni, B.; Lin, H.; Zhang, M.; Li, X.; Yin, X.; Qu, C.; Ni, J. Traditional usages, botany, phytochemistry, pharmacology and toxicology of Polygonum multiflorum thunb.: A review. J. Ethnopharmacol. 2015, 159, 158–183. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef] [PubMed]
- Eddie, W.; Shulkina, T.; Gaskin, J.; Haberle, R.; Jansen, R. Phylogeny of Campanulaceae s. Str. inferred from ITS sequences of nuclear ribosomal DNA. Ann. Mo. Bot. Gard. 2003, 90, 554–575. [Google Scholar] [CrossRef]
- Wendling, B.M.; Galbreath, K.E.; DeChaine, E.G. Resolving the evolutionary history of Campanula (Campanulaceae) in Western North America. PLoS ONE 2011, 6, e23559. [Google Scholar] [CrossRef] [PubMed]
- Crowl, A.A.; Mavrodiev, E.; Mansion, G.; Haberle, R.; Pistarino, A.; Kamari, G.; Phitos, D.; Borsch, T.; Cellinese, N. Phylogeny of Campanuloideae (Campanulaceae) with emphasis on the utility of nuclear pentatricopeptide repeat (PPR) genes. PLoS ONE 2014, 9, e94199. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.P.; Park, J.; Lee, Y.; Lee, M.; Park, S.G.; Uhm, Y.; Lee, J.; Kim, C.-K. accD nuclear transfer of Platycodon grandiflorum and the plastid of early Campanulaceae. BMC Genom. 2017, 18, 607. [Google Scholar] [CrossRef] [PubMed]
- Haberle, R.C.; Dang, A.; Lee, T.; Peñaflor, C.; Cortes-Burns, H.; Oestreich, A.; Raubeson, L.; Cellinese, N.; Edwards, E.J.; Kim, S.-T. Taxonomic and biogeographic implications of a phylogenetic analysis of the Campanulaceae based on three chloroplast genes. Taxon 2009, 58, 715–734. [Google Scholar]
- Byng, J.W.; Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Judd, W.S.; Mabberley, D.J.; Sennikov, A.N.; Soltis, D.E.; Soltis, P.S.; Stevens, P.F. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
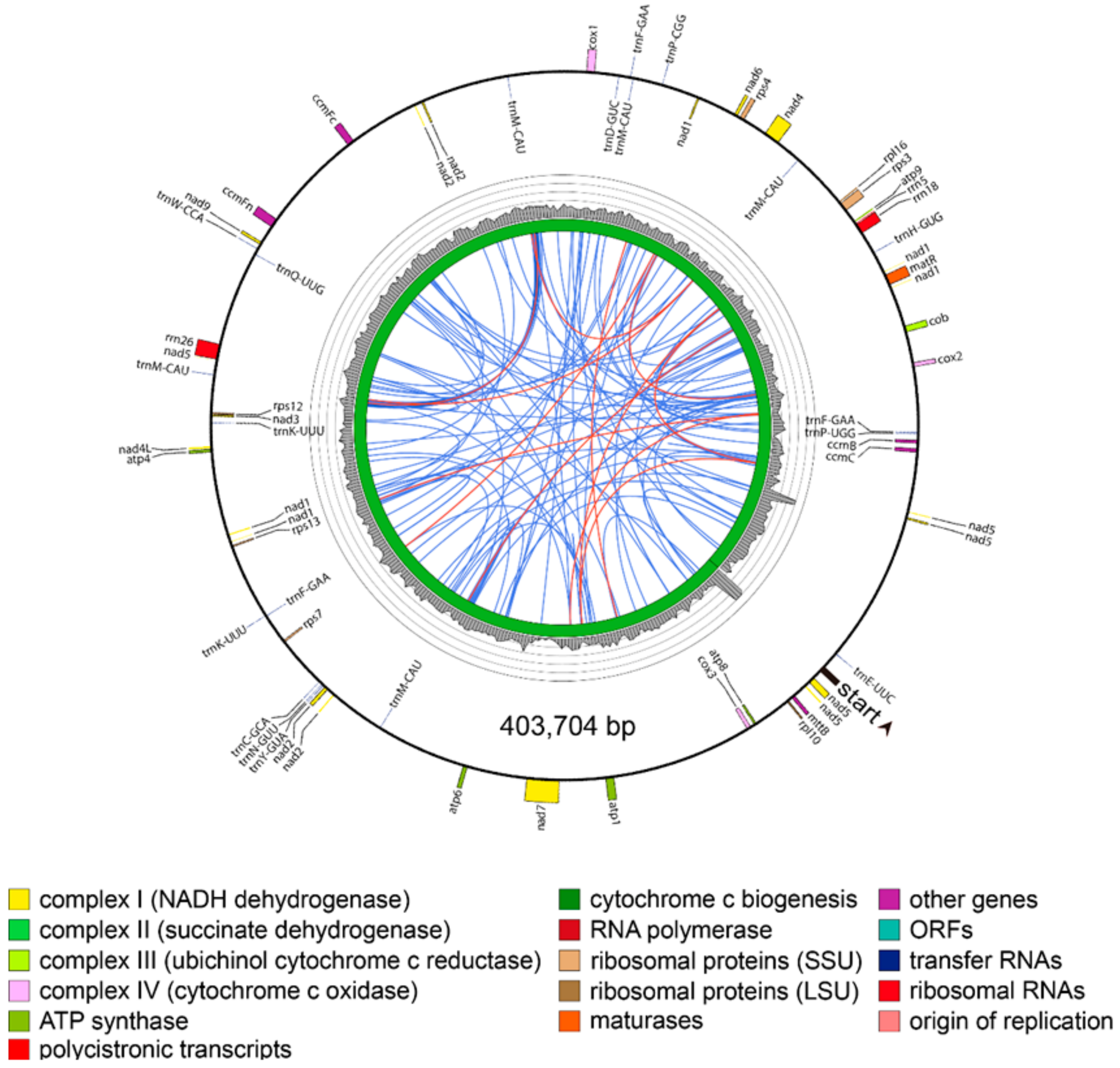{kind=link}
{kind=link}
| Category 1 | Common Genes | Unique Genes | |||
|---|---|---|---|---|---|
| P. Grandiflorus | C. Lanceolata | H. Annuus | |||
| (Master) | Minor | ||||
| Complex I | nad4L, nad9 | nad1–4, nad6, nad7 | nad2, nad4, nad7 | nad1–7 | nad3, nad5–6 |
| Complex II | sdh3, sdh4 | sdh3, sdh4 | |||
| Complex III | cob | ||||
| Complex IV | cox3 | cox1, cox2 | cox2 | cox1, cox2 | cox1 |
| Complex V | atp4, atp6, atp8, atp9 | atp1 | atp1 | atp1 | |
| Cytochrome | ccmB, ccmC, ccmFc, ccmFn | ||||
| Large subunit | rpl10, rpl16 | rpl5 | |||
| Small subunit | rps13 | rps3, rps4, rps7, rps12 | rps3, rps7 | rps3, rps4, rps7, rps12 | rps4, rps12, rps13 |
| Maturase | matR | matR | matR | ||
| Transport protein | mttB | mttB | |||
| Ribosomal RNAs | rrn5, rrn18, rrn26 | ||||
| Transfer RNAs | trnC-GCA, trnE-UUC, trnF-GAA, trnH-GUG, trnM-CAU, trnP-UGG, trnQ-UUG, trnW-CCA, trnY-GUA | trnK-UUU, trnL-CAA | trnL-CAA | trnD-GUC, trnK-UUU, trnN-GUU, trnP-CGG | trnD-GUC, trnG-GCC, trnN-GUU, trnS-GCT |
| Total genes | 27 | 19 | 9 | 20 | 13 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-O.; Choi, J.-W.; Baek, J.-H.; Oh, J.-H.; Lee, S.-C.; Kim, C.-K. Assembly of the Mitochondrial Genome in the Campanulaceae Family Using Illumina Low-Coverage Sequencing. Genes 2018, 9, 383. https://doi.org/10.3390/genes9080383
Lee H-O, Choi J-W, Baek J-H, Oh J-H, Lee S-C, Kim C-K. Assembly of the Mitochondrial Genome in the Campanulaceae Family Using Illumina Low-Coverage Sequencing. Genes. 2018; 9(8):383. https://doi.org/10.3390/genes9080383
Chicago/Turabian StyleLee, Hyun-Oh, Ji-Weon Choi, Jeong-Ho Baek, Jae-Hyeon Oh, Sang-Choon Lee, and Chang-Kug Kim. 2018. "Assembly of the Mitochondrial Genome in the Campanulaceae Family Using Illumina Low-Coverage Sequencing" Genes 9, no. 8: 383. https://doi.org/10.3390/genes9080383
APA StyleLee, H. -O., Choi, J. -W., Baek, J. -H., Oh, J. -H., Lee, S. -C., & Kim, C. -K. (2018). Assembly of the Mitochondrial Genome in the Campanulaceae Family Using Illumina Low-Coverage Sequencing. Genes, 9(8), 383. https://doi.org/10.3390/genes9080383