Long-Term Observation of Atmospheric Speciated Mercury during 2007–2018 at Cape Hedo, Okinawa, Japan

Abstract
:1. Introduction
2. Materials and Methods
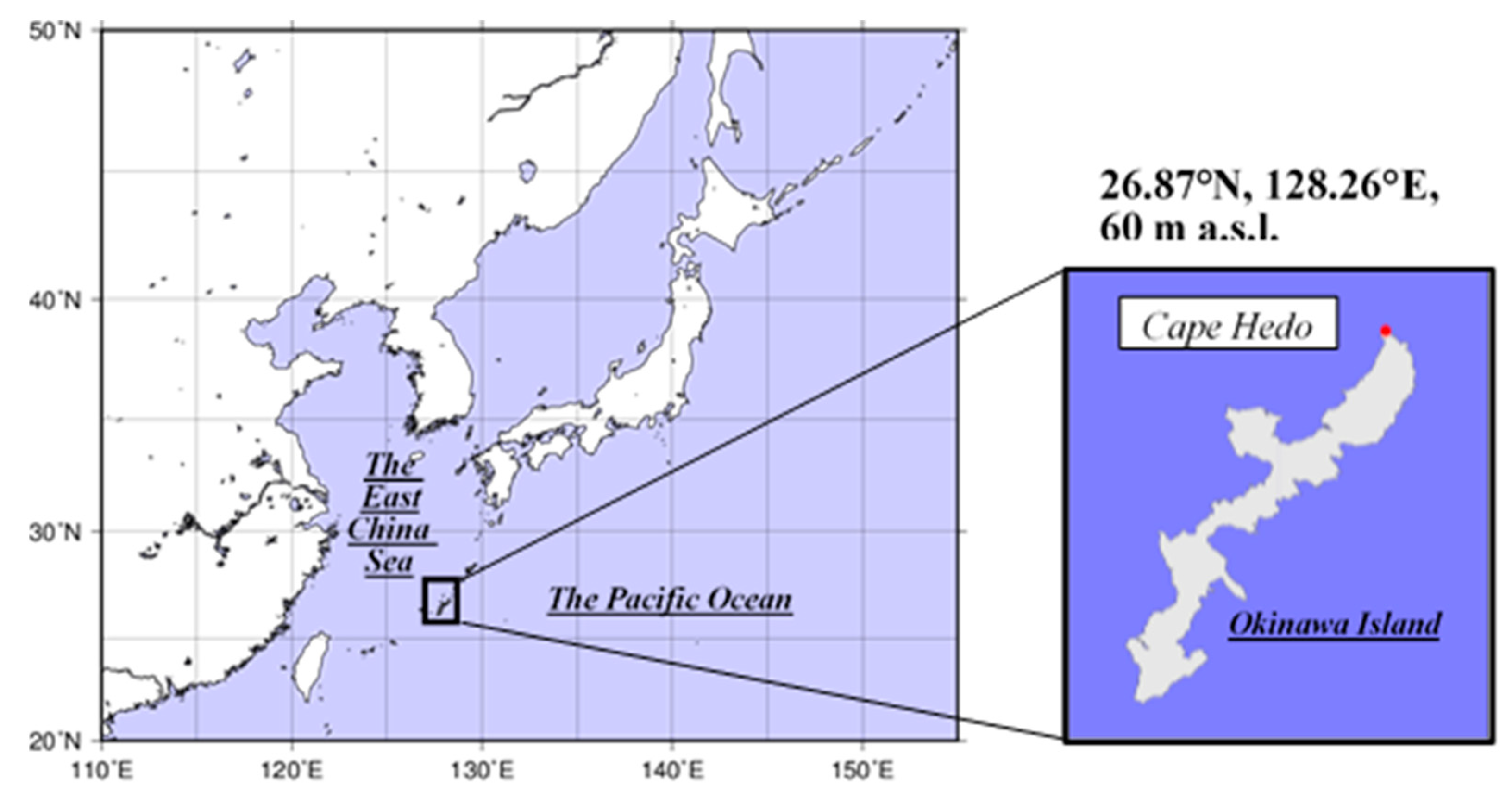
2.1. Site Information
2.2. Atmospheric Speciated Hg Measurements
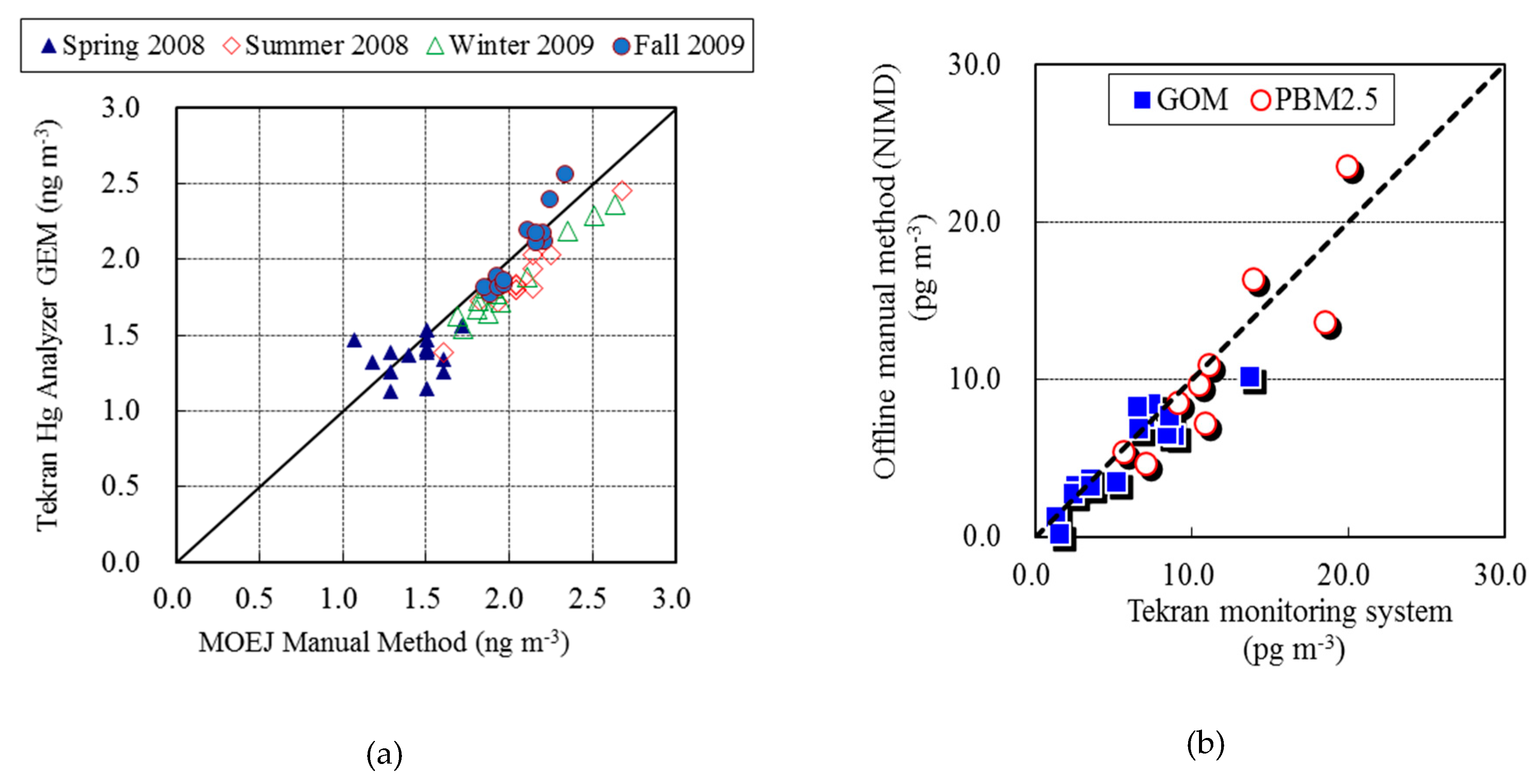
2.3. QA/QC Program for Speciated Hg
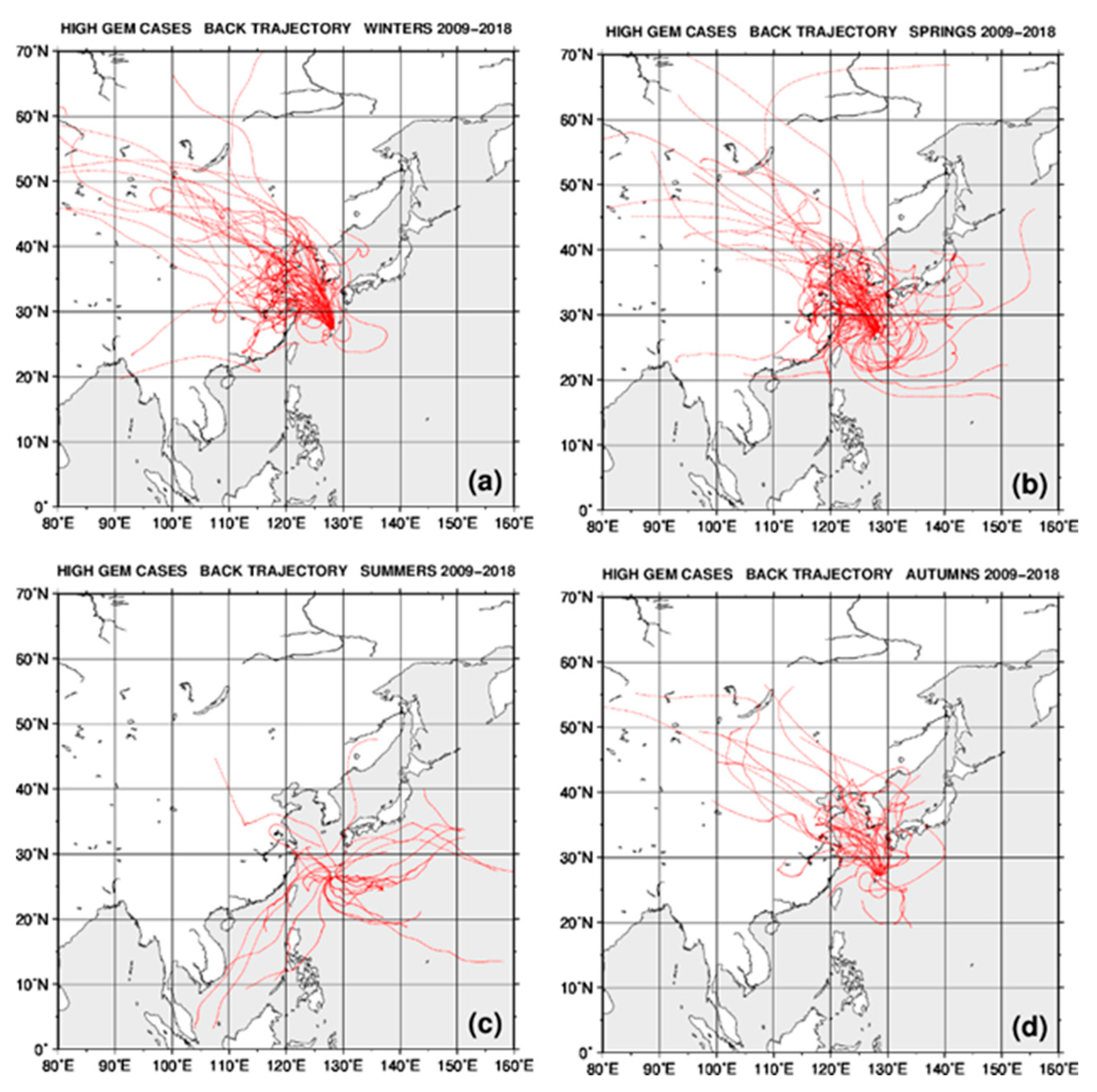
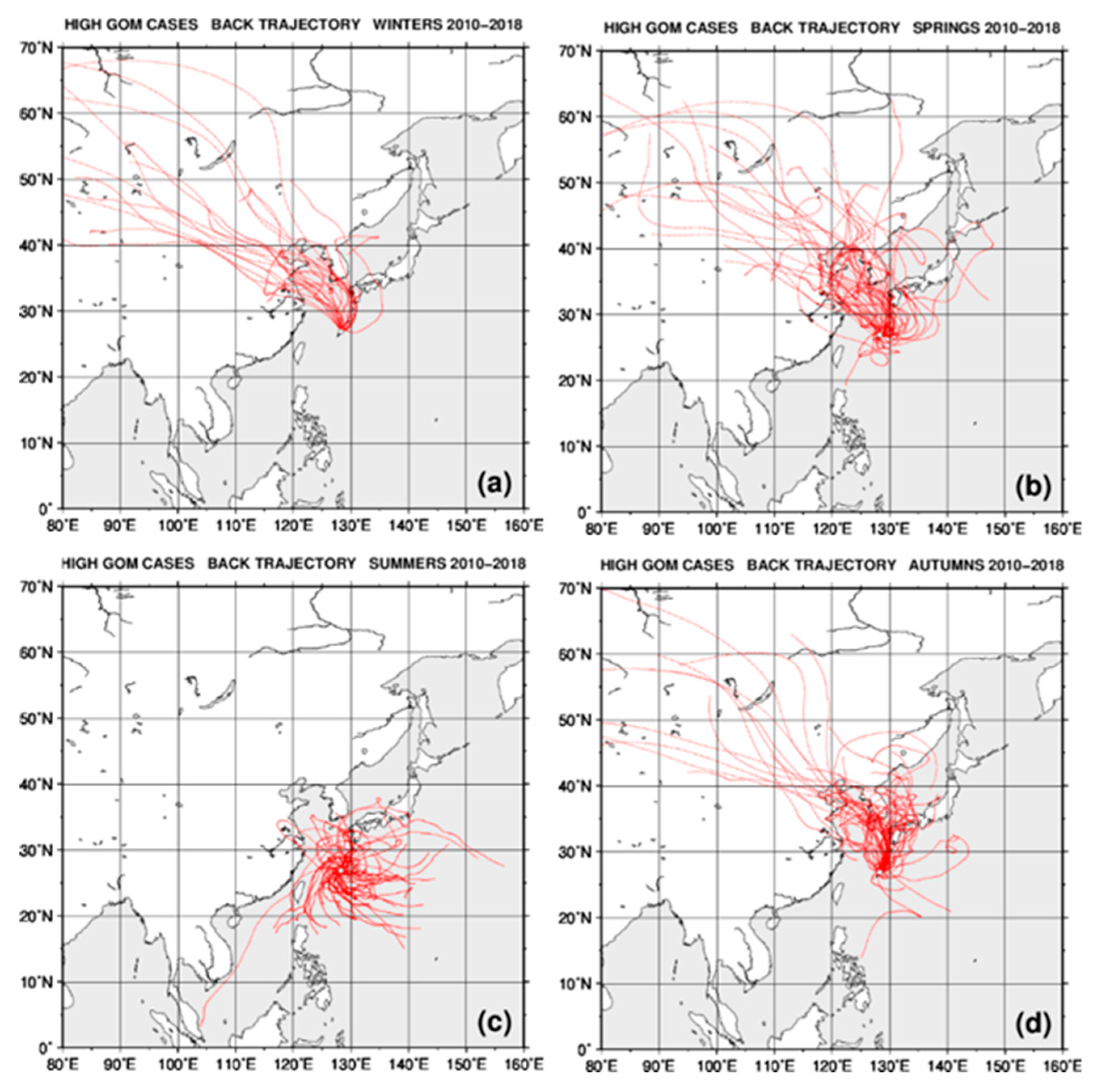
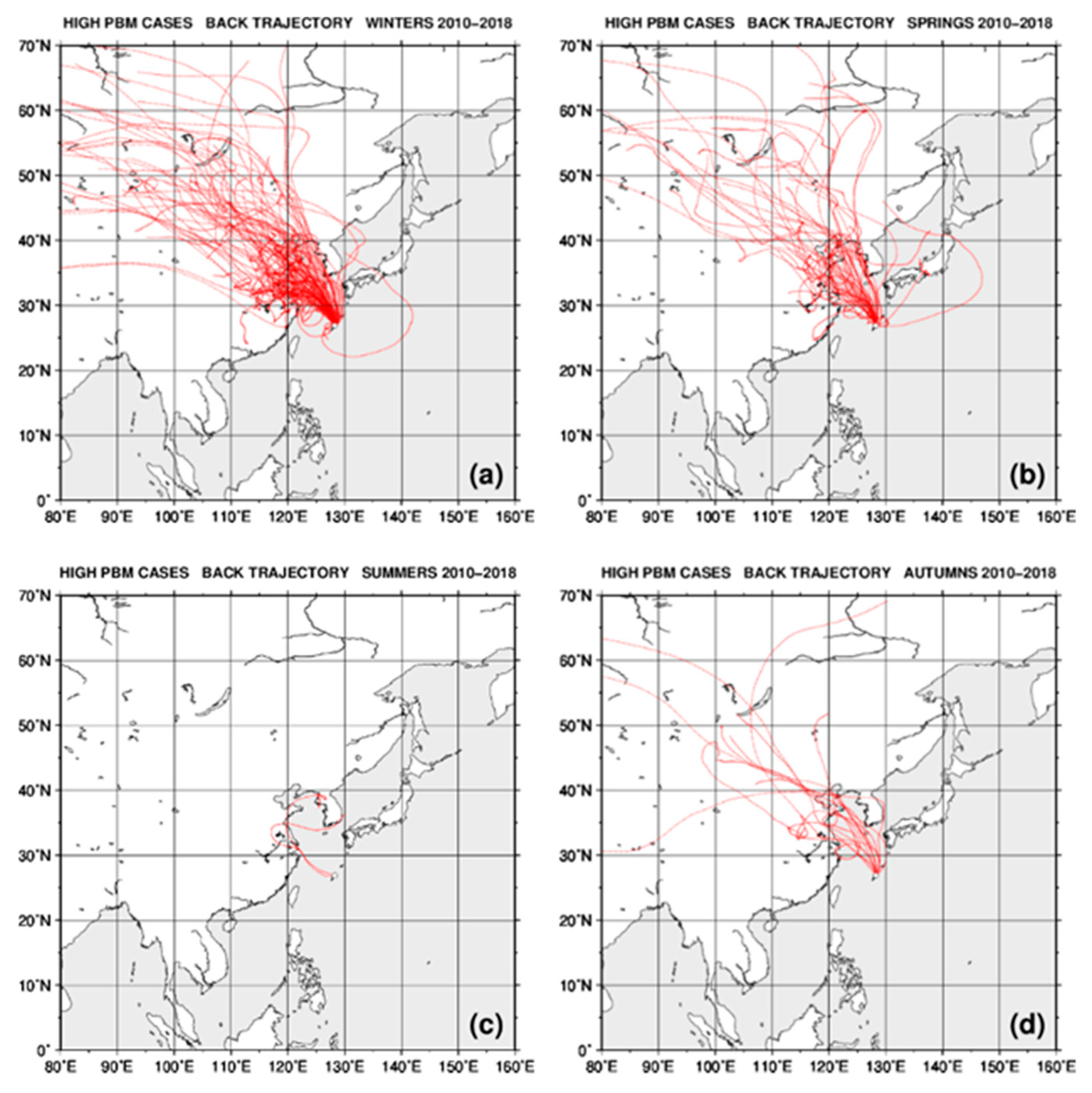
2.4. Back Trajectory Analysis
3. Results and Discussion
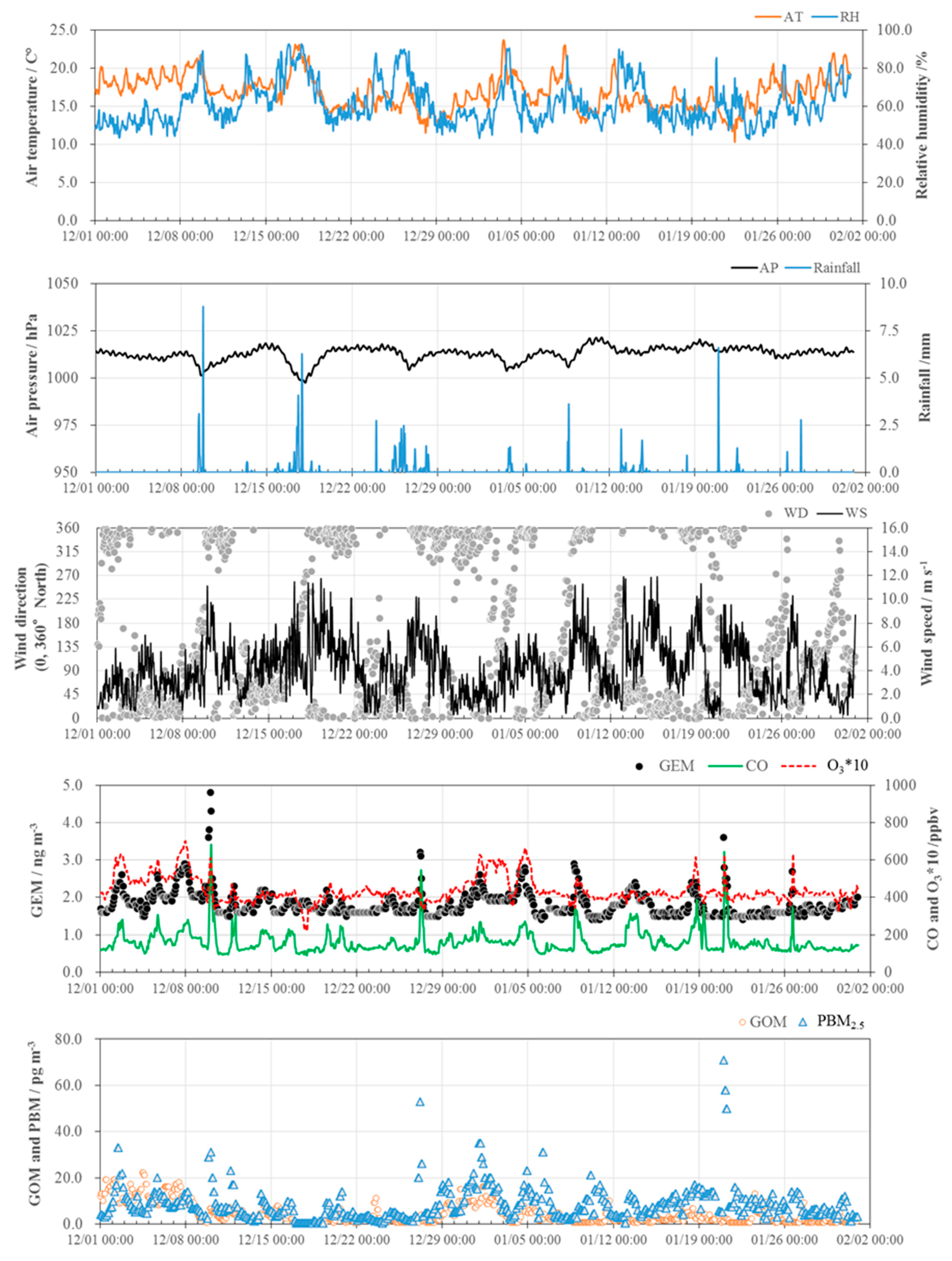
3.1. Concentrations of GEM, GOM, and PBM2.5
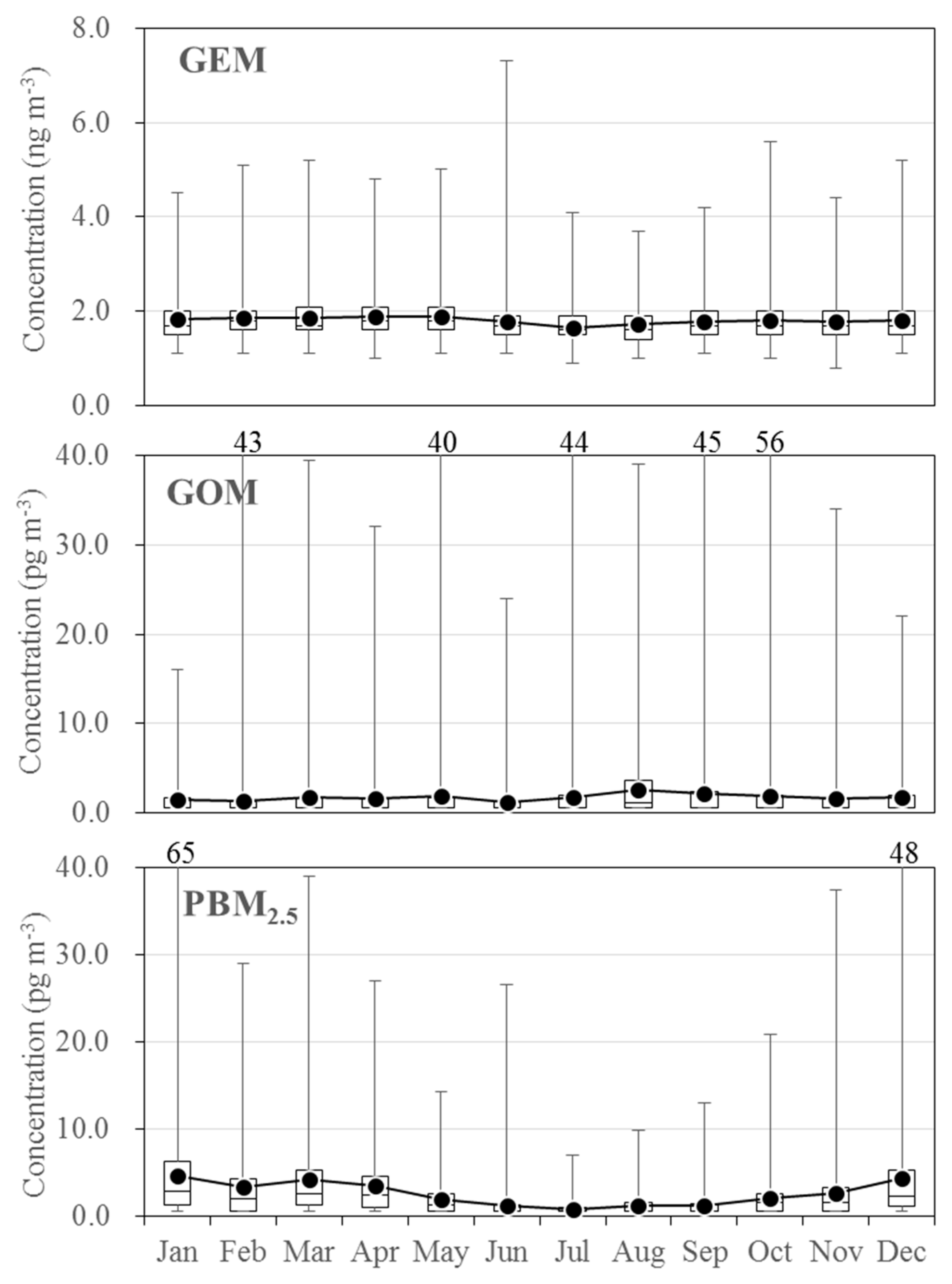
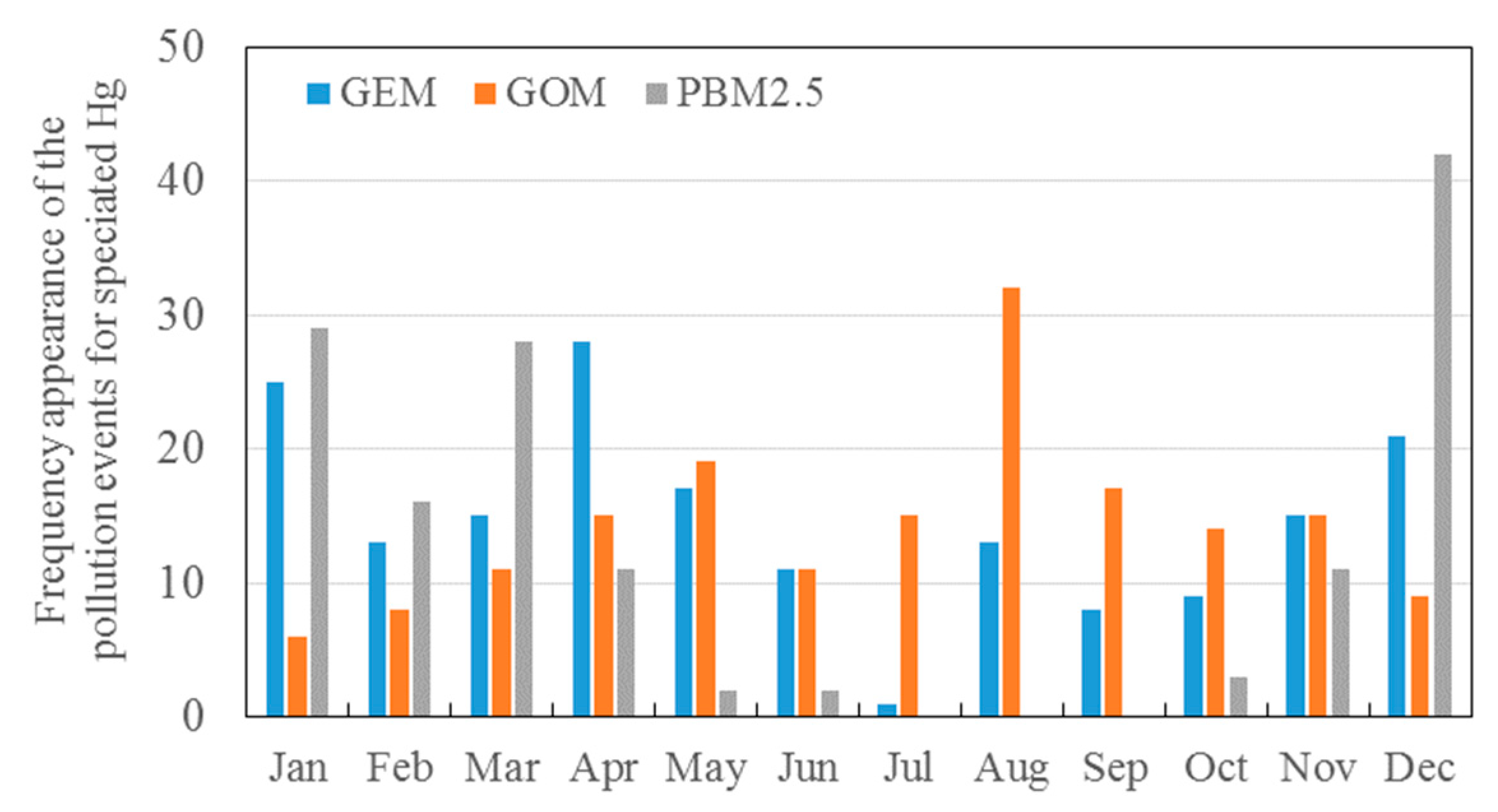
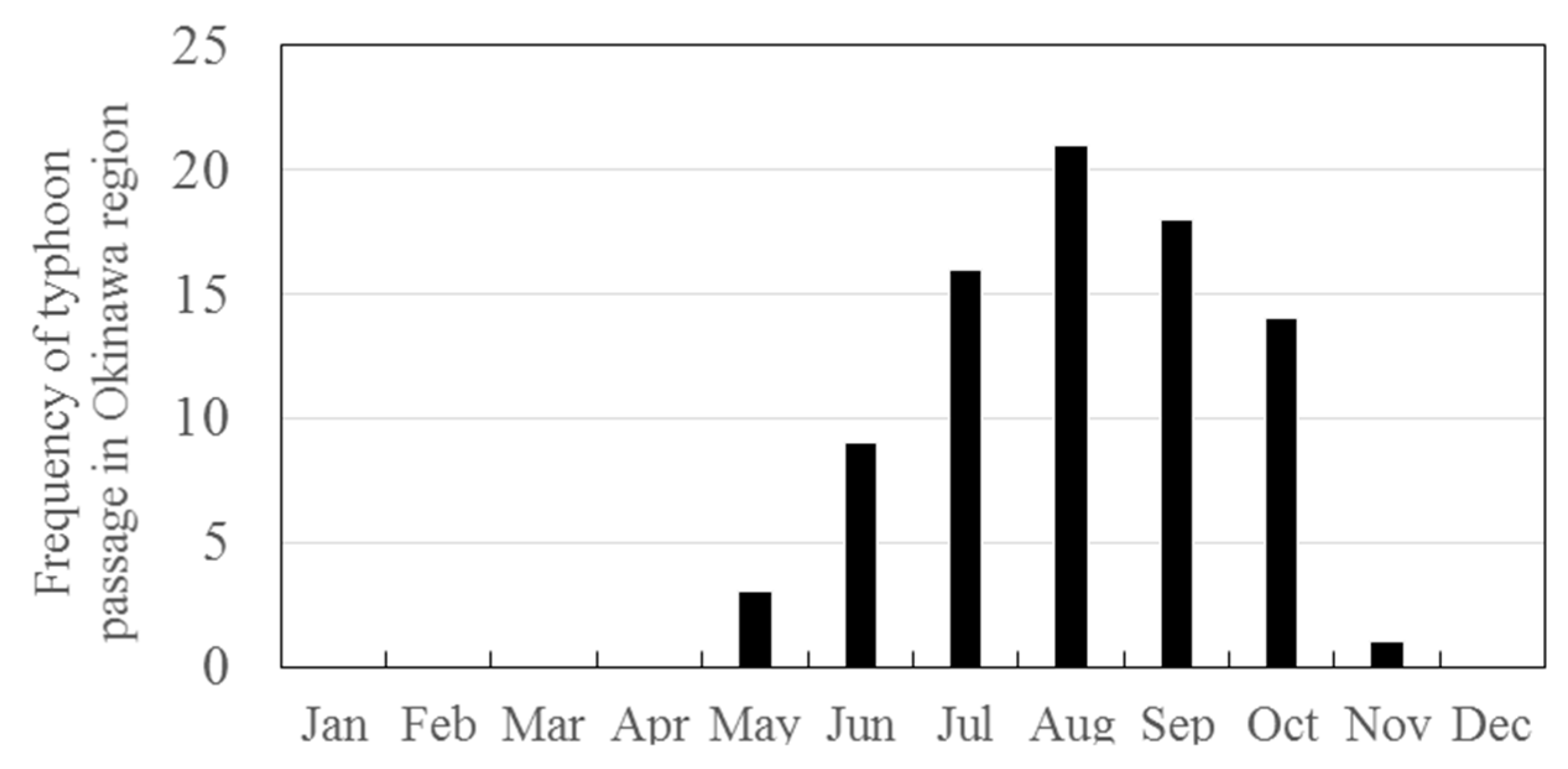
3.2. Seasonal Variations in the Concentrations of GEM, GOM, and PBM2.5
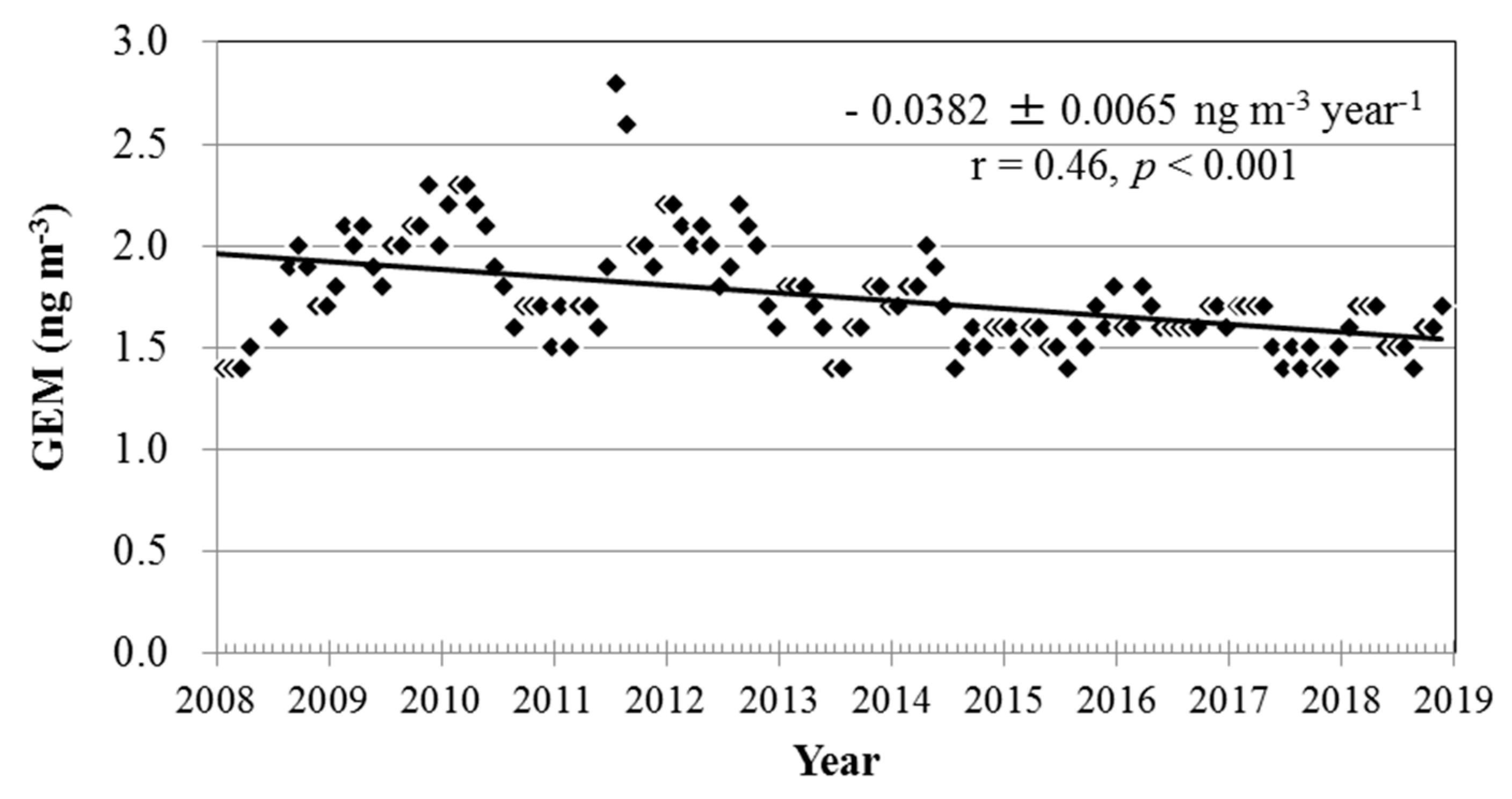
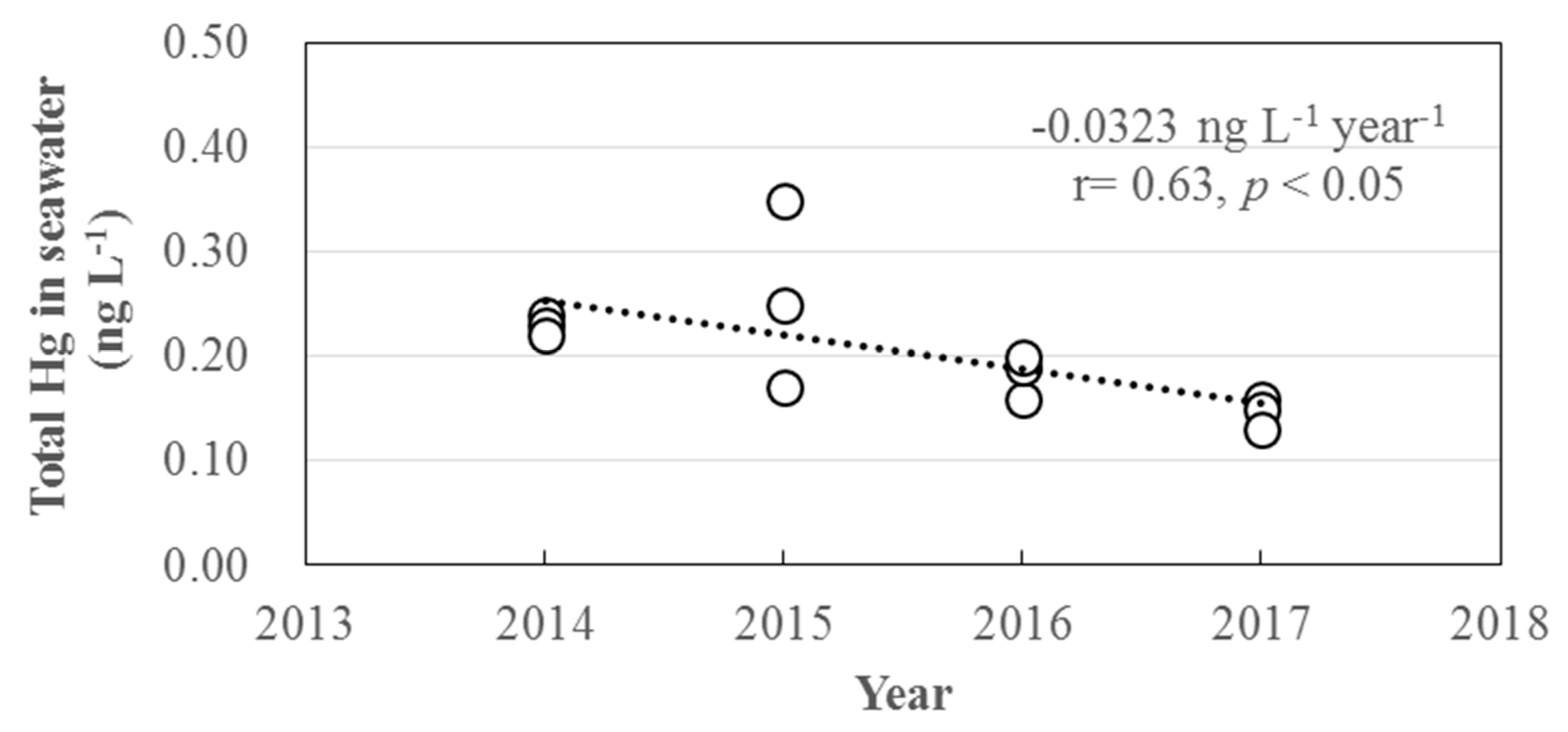
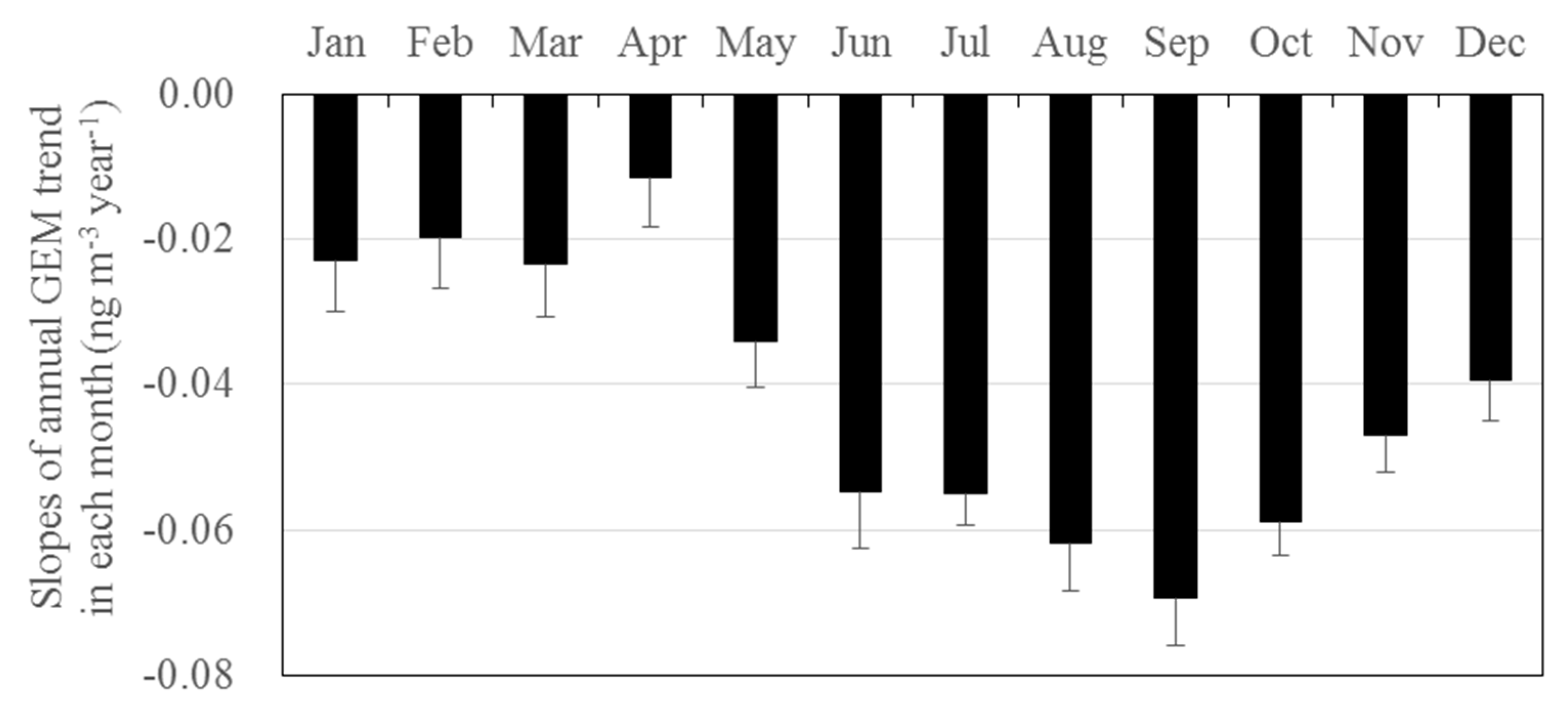
3.3. Long-Term Trends of Atmospheric GEM, GOM, and PBM2.5 Concentrations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schroeder, W.H.; Munthe, J. Atmospheric mercury-an overview. Atmos. Environ. 1998, 32, 809–822. [Google Scholar] [CrossRef]
- Lindberg, S.E.; Stratton, W.J. Atmospheric mercury speciation: Concentration and behavior of reactive gaseous mercury in ambient air. Environ. Sci. Technol. 1998, 32, 49–57. [Google Scholar] [CrossRef]
- Rutter, A.P.; Snyder, D.C.; Stone, E.A.; Schauer, J.J.; Gonzalez-Abraham, R.; Molina, L.T.; Marquez, C.; Cardenas, B.; de Foy, B. In situ measurements of speciated atmospheric mercury and the identification of source regions in the Mexico City Metropolitan Area. Atmos. Chem. Phys. 2009, 9, 207–220. [Google Scholar] [CrossRef] [Green Version]
- United Nations Environmenta Programme (UNEP). The Global Atmospheric Mercury Assessment: Sources, Emissions and Transport; UNEP Chemicals Branch, DTIE: Geneva, Switzerland, 2013. [Google Scholar]
- Chand, D.; Jaffe, D.; Prestbo, E.; Swartzendruber, P.C.; Hafner, W.; Weiss-Penzias, P.; Kato, S.; Takami, A.; Hatakeyama, S.; Kaiji, Y. Reactive and particulate mercury in the Asian marine boundary layer. Atmos. Environ. 2008, 42, 7988–7996. [Google Scholar] [CrossRef]
- Lin, C.-J.; Pehkonen, S.O. Two-phase model of mercury chemistry in the atmosphere. Atmos. Environ. 1998, 32, 2543–2558. [Google Scholar] [CrossRef]
- Hedgecock, I.M.; Pirrone, N. Chasing Quicksilver: Modeling the atmospheric lifetime of Hg0(g) in the marine boundary layer at various latitudes. Environ. Sci. Technol. 2004, 38, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Rutter, A.P.; Schauer, J.J. The impact of aerosol composition on the particle to gas partitioning of reactive mercury. Environ. Sci. Technol. 2007, 41, 3934–3939. [Google Scholar] [CrossRef] [PubMed]
- Rutter, A.P.; Schauer, J.J. The effect of temperature on the gas-particle partitioning of reactive mercury in atmospheric aerosols. Atmos. Environ. 2007, 39, 8647–8657. [Google Scholar] [CrossRef]
- Sakata, M.; Marumoto, K. Formation of atmospheric particulate mercury in the Tokyo metropolitan area. Atmos. Environ. 2002, 36, 239–246. [Google Scholar] [CrossRef]
- Landis, M.S.; Stevens, R.K.; Schaedlich, F.; Prestbo, E.M. Development and characterization of an annular denuder methodology for the measurement of divalent inorganic reactive gaseous mercury in ambient air. Environ. Sci. Technol. 2002, 36, 3000–3009. [Google Scholar] [CrossRef]
- Zhang, L.; Wright, L.P.; Blanchard, P. A review of current knowledge concerning dry deposition of atmospheric mercury. Atmos. Environ. 2009, 43, 5853–5864. [Google Scholar] [CrossRef]
- Sprovieri, F.; Pirrone, N.; Ebinghaus, R.; Kock, H.; Dommergue, A. A review of worldwide atmospheric mercury measurements. Atmos. Chem. Phys. 2010, 10, 8245–8265. [Google Scholar] [CrossRef] [Green Version]
- Sprovieri, F.; Pirrone, N.; Bencardino, M.; D’Amorel, F.; Carbone, F.; Cinnirella, S.; Mannarino, V.; Landis, M.; Ebinghaus, R.; Weigelt, A.; et al. Atmospheric mercury concentrations observed at ground-based monitoring sites globally distributed in the framework of the GMOS network. Atmos. Chem. Phys. 2016, 16, 11915–11935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.W.; Zhang, H.; Yu, B.; Wang, X.; Lin, C.-J.; Feng, X.B. Observations of atmospheric mercury in China: A critical review. Atmos. Chem. Phys. 2015, 15, 9455–9476. [Google Scholar] [CrossRef]
- Ebinghaus, R.; Jenning, S.G.; Kock, H.H.; Derwent, R.G.; Manning, A.J.; Spain, T.G. Decreasing trends in total gaseous mercury observations in baseline air at Mace Head, Ireland from 1996 to 2009. Atmos. Environ. 2011, 45, 3475–3480. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, A.; Ebinghaus, R.; Manning, A.J.; Derwent, R.G.; Simmonds, P.G.; Spain, T.G.; Jennings, S.G.; Slemr, F. Analysis and interpretation of 18 years of mercury observations since 1996 at Mace Head, Ireland. Atmos. Environ. 2015, 100, 85–93. [Google Scholar] [CrossRef]
- Slemr, F.; Brunke, E.-G.; Ebinghaus, R.; Temme, C.; Munthe, J.; Wangberg, I.; Schroeder, W.; Steffen, A.; Berg, T. Worldwide trend of atmospheric mercury since 1977. Geophys. Res. Lett. 2003, 30, 1516. [Google Scholar] [CrossRef]
- Cole, A.S.; Steffen, A. Trends in long-term gaseous mercury observations in the Arctic and effects of temperature and other atmospheric conditions. Atmos. Chem. Phys. 2010, 10, 4661–4672. [Google Scholar] [CrossRef] [Green Version]
- Slemr, F.; Brunke, E.-G.; Labuschagne, C.; Ebinghaus, R. Total gaseous mercury concentrations at the Cape Point GAW station and their seasonality. Geophys. Res. Lett. 2008, 35, 11. [Google Scholar] [CrossRef]
- Martin, L.G.; Labuschagne, C.; Brunke, E.-G.; Weigelt, A.; Ebinghaus, R.; Slemr, F. Trend of atmospheric mercury concentrations at Cape Point for 1995–2004 and since 2007. Atmos. Chem. Phys. 2017, 17, 2393–2399. [Google Scholar] [CrossRef]
- Berg, T.; Pfaffhuber, A.; Colu, A.S.; Engelsen, O.; Steffen, A. Ten-year trends in atmospheric mercury concentrations, meteorological effects and climate variables at Zeppelin, Ny-Alesund. Atmos. Chem. Phys. 2013, 13, 6575–6586. [Google Scholar] [CrossRef]
- Streets, D.G.; Devane, M.K.; Lu, Z.; Bond, T.C.; Sunderland, E.M.; Jacob, D.J. All-time releases of mercury to the atmosphere from human activities. Environ. Sci. Technol. 2011, 45, 10485–10491. [Google Scholar] [CrossRef] [PubMed]
- Slemr, F.; Brunke, E.-G.; Ebinghaus, R.; Kuss, J. Worldwide trend of atmospheric mercury since 1995. Atmos. Chem. Phys. 2011, 11, 4779–4787. [Google Scholar] [CrossRef] [Green Version]
- Soerensen, A.L.; Jacob, D.J.; Streets, D.G.; Witt, M.L.I.; Ebinghaus, R.; Mason, R.P.; Andersson, M.A.; Sunderland, E.M. Multi-decadal decline of mercury in the North Atlantic atmosphere explained by changing subsurface seawater concentrations. Geophys. Res. Lett. 2012, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jacob, D.J.; Horowitz, H.M.; Chen, L.; Amos, H.M.; Krabbenhort, D.P.; Slemr, F.; StLouis, V.L.; Sunderland, E.M. Observed decrease in atmospheric mercury explained by global decline in anthropogenic emissions. Proc. Natl. Acad. Sci. USA 2016, 113, 526–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streets, D.G.; Horowitz, H.M.; Jacob, D.J.; Lu, Z.; Levin, L.; Schure, A.; Sunderland, E.M. Total mercury released to the environment by human activities. Environ. Sci. Technol. 2017, 51, 5969–5977. [Google Scholar] [CrossRef]
- Sheu, G.-R.; Lin, N.-H.; Wang, J.-L.; Lee, C.-T.; Ou Yang, C.-F.; Wang, S.-H. Temporal distribution and potential sources of atmospheric mercury measured at a high-elevation background station in Taiwan. Atmos. Environ. 2010, 44, 2393–2400. [Google Scholar] [CrossRef]
- Lee, S.; Akimoto, H.; Nakane, H.; Kurnosenko, S.; Kinjo, Y. Lower tropospheric ozone trend observed in 1989–1997 at Okinawa, Japan. Geophys. Res. Lett. 1998, 25, 1637–1640. [Google Scholar] [CrossRef]
- Suthawaree, J.; Kato, S.; Takami, A.; Kadena, H.; Toguchi, M.; Yogi, K.; Hatakeyama, S.; Kaiji, Y. Observation of ozone and carbon monoxide at Capr Hedo, Japan: Seasonal variation and influence of long-range transport. Atmos. Environ. 2008, 42, 2971–2981. [Google Scholar] [CrossRef]
- Handa, D.; Nakajima, H.; Arakaki, T.; Kumata, H.; Shibata, Y.; Uchida, M. Radiocarbon analysis of BC and OC in PM10 aerosols at Cape Hedo, Okinawa, Japan, during long-range transport events from East Asian countries. Nucl. Instrum. Methods Phys. B 2010, 268, 1125–1128. [Google Scholar] [CrossRef]
- Verma, R.L.; Kondo, Y.; Oshima, N.; Matsui, H.; Kita, K.; Sahu, L.K.; Kato, S.; Kaiji, Y.; Takami, A.; Miyakawa, T. Seasonal variation of the transport of black carbon and carbon monoxide from the Asian Continent to the western Pacific in the boundary layer. J. Geophys. Res. Atmos. 2011, 116, D21307. [Google Scholar] [CrossRef]
- Matsui, H.; Koike, M.; Kondo, Y.; Oshima, N.; Moteki, N.; Kanaya, Y.; Takami, A.; Irwin, M. Seasonal variations of Asian black carbon outflow to the Pacific: Contribution from anthropogenic sources in China and biomass burning sources in Siberia and Southeast Asia. J. Geophys. Res. 2013, 118, 9948–9996. [Google Scholar] [CrossRef]
- Sato, K.; Li, H.; Tanaka, Y.; Ogawa, S.; Iwasaki, Y.; Takami, A.; Hatakeyama, S. Long-range transport of particulate polycyclic aromatic hydrocarbons at Cape Hedo remote island site in the East China Sea between 2005 and 2008. J. Atmos. Chem. 2008, 61, 243–257. [Google Scholar] [CrossRef] [Green Version]
- Pani, S.K.; Lee, C.-T.; Chou, C.-K.; Shimada, K.; Hatakeyama, S.; Takami, A.; Wang, S.-H.; Lin, N.-H. Chemical characterization of wintertime aerosols over islands and mountains in East Asia: Impacts of the continental Asian outflows. Aerosol. Air Qual. Res. 2017, 17, 3006–3036. [Google Scholar] [CrossRef]
- Shimada, K.; Yang, X.; Araki, Y.; Yoshino, A.; Takami, A.; Chen, X.; Meng, F.; Hatakeyama, S. Concentrations of metallic elements in long-range-transported aerosols measured simultaneously at three coastal sites in China and Japan. J. Atmos. Chem. 2018, 75, 123–139. [Google Scholar] [CrossRef]
- Itoh, A.; Oshiro, Y.; Azechi, S.; Somada, Y.; Handa, D.; Miyagi, Y.; Nakano, K.; Tanahara, A.; Arakaki, T. Long-term monitoring of metal elements in total suspended particle aerosols simultaneously collected at three islands in Okinawa, Japan. Asian J. Atmos. Environ. 2018, 12, 326–337. [Google Scholar] [CrossRef]
- Jaffe, D.; Prestbo, E.; Swartzendruber, P.; Weiss-Penzias, P.; Kato, S.; Takami, A.; Hatakeyama, S.; Kaiji, Y. Export of atmospheric mercury from Asia. Atmos. Environ. 2005, 39, 3029–3038. [Google Scholar] [CrossRef]
- Tekran Inc. MODEL 2537A Mercury Vapor Analyzer User Manual; Tekran Inc.: Toronto, ON, Canada, 2002. [Google Scholar]
- Ministry of the Environment, Japan. Manual Method of Hazardous Air Pollutants, Air Environmental Division of the Environmental Management Bureau for Water and Air Environmental Fields; MOEJ: Tokyo, Japan, 2011. (In Japanese)
- Draxler, R.R.; Rolph, G.D. HYSPLIT (Hybrid. Single-Particle Lagrangian Integrated Trajectory) Model. Access via NOAA Arl Ready Website; NOAA Air Resources Laboratory: Silver Springs, MD, USA, 2010. [Google Scholar]
- Kim, K.-H.; Brown, R.J.C.; Kwon, E.; Kim, I.-S.; Sohn, J.-R. Atmospheric mercury at an urban station in Korea across three decades. Atmos. Environ. 2016, 131, 124–132. [Google Scholar] [CrossRef]
- Lee, G.-S.; Kim, P.-R.; Han, Y.-J.; Holsen, T.M.; Seo, Y.-S.; Yi, S.-M. Atmospheric speciated mercury concentrations on the island between China and Korea: Sources and transport pathways. Atmos. Chem. Phys. 2016, 16, 4119–4133. [Google Scholar] [CrossRef]
- Marumoto, K.; Hayashi, M.; Takami, A. Atmospheric mercury concentrations at two sites in the Kyushu Islands, Japan, and evidence of long-range transport from East Asia. Atmos. Environ. 2015, 117, 147–155. [Google Scholar] [CrossRef]
- Qin, X.; Wang, X.; Shi, Y.; Yu, G.; Zhao, N.; Lin, Y.; Fu, Q.; Wang, D.; Xie, Z.; Deng, C.; et al. Characteristics of atmospheric mercury in a suburban area of east China: Sources, formation mechanisms, and regional transport. Atmos. Chem. Phys. 2019, 19, 5923–5940. [Google Scholar] [CrossRef]
- Slemr, F.; Angot, H.; Dommergue, A.; magand, O.; Barret, M.; Weigelt, A.; Ebinghaus, R.; Brunke, E.-G.; Plaffhuber, K.A.; Edwards, G.; et al. Comparison of mercury concentrations measured at several sites in the Southern Hemisphere. Atmos. Chem. Phys. 2015, 15, 3125–3133. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ci, Z.; Wang, Z.; Zhang, X.; Guo, J. Speciated atmospheric mercury in the marine boundary layer of the Bohai Sea and Yellow Sea. Atmos. Environ. 2016, 131, 360–370. [Google Scholar] [CrossRef]
- Kim, S.-H.; Han, Y.-J.; Holsen, T.M.; Yi, S.-M. Characteristics of atmospheric speciated mercury concentrations (TGM, Hg(II) and Hg(p)) in Seoul, Korea. Atmos. Environ. 2009, 43, 3267–3274. [Google Scholar] [CrossRef]
- Fu, X.; Feng, X.; Sommar, J.; Wang, S. A review of studies on atmospheric mercury in China. Sci. Total Environ. 2012, 421–422, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, H.M.; Jacob, D.J.; Zhang, Y.; Dibble, T.S.; Slemr, F.; Amos, H.M.; Schmidt, J.A.; Corbitt, E.S.; Marais, E.A.; Sunderland, E.M. A new mechanism for atmospheric mercury redox chemistry: Implications for the global mercury budget. Atmos. Chem. Phys. 2017, 17, 6353–6371. [Google Scholar] [CrossRef]
- Holmes, C.; Jacob, D.; Mason, R.; Jaffe, D. Sources and deposition of reactive gaseous mercury in the marine atmosphere. Atmos. Environ. 2009, 43, 2278–2285. [Google Scholar] [CrossRef] [Green Version]
- Japanese Meteorological Agency. 2019. Available online: https://www.jma.go.jp/fcd/yoho/typhoon/index.html (accessed on 20 May 2019).
- Kaneyasu, N.; Takeuchi, K.; Hayashi, M.; Fujita, S.; Uno, I.; Sasaki, H. Outflow patterns of pollutants from East Asia to the North Pacific in the winter monsoon. J. Geophys. Res. Atmos. 2000, 105, 17361–17377. [Google Scholar] [CrossRef]
- Kaneyasu, N.; Yamamoto, S.; Sato, K.; Takami, A.; Hayashi, M.; Hara, K.; Kawamoto, K.; Okuda, T.; Hatakeyama, S. Impact of long-range transport of aerosols on the PM2.5 composition at a major metropolitan area in the northern Kyushu area of Japan. Atmos. Environ. 2014, 97, 416–425. [Google Scholar] [CrossRef]
- Japan Coast Guard. Report of Marine Pollution Surveys No.42-No.45, Hydrographical and Oceanographic Department; Japan Coast Guard: Tokyo, Japan, 2016–2019. (In Japanese)
- U.S. EPA. Method 1631, Revision E: Mercury in Water by Oxidation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectrometry; US Environmental Protection Agency: Washington, DC, USA, 2002; EPA-821-R-02-019.
- Obrist, D.; Kirk, J.L.; Zhang, L.; Sunderland, E.M.; Jiskra, M.; Selin, N.E. A review of global environmental mercury processes in response to human and natural perturbations: Changes of emissions, climate, and land use. Ambio 2018, 47, 116–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Wang, S.; Li, G.; Liang, S.; Lin, C.-J.; Wang, Y.; Cai, S.; Liu, K.; Hao, J. Temporal trend ans spatial distribution of speciated atmospheric mercury emissions in China during 1978–2014. Environ. Sci. Technol. 2016, 50, 13428–13435. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, C.-J.; Feng, X.; Yuan, W.; Fu, X.; Zhang, H.; Wu, Q.; Wang, S. Assessment of regional mercury deposition and emission outflow in Mainland China. J. Geophys. Res. Atmos. 2018, 123, 9868–9890. [Google Scholar] [CrossRef]
- Streets, D.G.; Horowitz, H.M.; Lu, Z.; Levin, L.; Thackray, C.P.; Sunderland, E.M. Global and regional trends in mercury emissions and concentrations, 2010–2015. Atmos. Environ. 2019, 201, 417–427. [Google Scholar] [CrossRef]
- Swartzendruber, P.C.; Jaffe, D.A.; Finley, B. Improved fluorescence peak integration in the Tekran 2537 for applications with sub-optical sample loadings. Atmos. Environ. 2009, 43, 3648–3651. [Google Scholar]
- Slemr, F.; Weigelt, A.; Ebinghaus, R.; Kock, H.H.; Bödewadt, J.; Brenninkmeijer, C.A.M.; Rauthe-Schöch, A.; Weber, S.; Hermann, M.; Becker, J.; et al. Atmospheric mercury measurements on board the CARIBIC passenger aircraft. Atmos. Meas. Tech. 2016, 9, 2291–2302. [Google Scholar] [CrossRef]
- Ambrose, J.L. Improved methods for signal processing in measurements of mercury by Tekran® 2537A and 2537B instruments. Atmos. Meas. Tech. 2017, 10, 5063–5073. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEM (ng m−3) | GOM (pg m−3) | PBM2.5 (pg m−3) | |
|---|---|---|---|
| N | 53,840 | 22,071 | 22,058 |
| Mean ± S.D. | 1.81 ± 0.43 | 1.7 ± 2.9 | 2.6 ± 3.6 |
| Minimum | 0.8 | 0.5 | 0.5 |
| 25 percentile | 1.5 | 0.5 | 0.5 |
| Median | 1.7 | 0.5 | 1.4 |
| 75 percentile | 2.0 | 1.8 | 3.1 |
| Maximum | 7.3 | 58 | 71 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marumoto, K.; Suzuki, N.; Shibata, Y.; Takeuchi, A.; Takami, A.; Fukuzaki, N.; Kawamoto, K.; Mizohata, A.; Kato, S.; Yamamoto, T.; et al. Long-Term Observation of Atmospheric Speciated Mercury during 2007–2018 at Cape Hedo, Okinawa, Japan. Atmosphere 2019, 10, 362. https://doi.org/10.3390/atmos10070362
Marumoto K, Suzuki N, Shibata Y, Takeuchi A, Takami A, Fukuzaki N, Kawamoto K, Mizohata A, Kato S, Yamamoto T, et al. Long-Term Observation of Atmospheric Speciated Mercury during 2007–2018 at Cape Hedo, Okinawa, Japan. Atmosphere. 2019; 10(7):362. https://doi.org/10.3390/atmos10070362
Chicago/Turabian StyleMarumoto, Kohji, Noriyuki Suzuki, Yasuyuki Shibata, Akinori Takeuchi, Akinori Takami, Norio Fukuzaki, Kazuaki Kawamoto, Akira Mizohata, Shungo Kato, Takashi Yamamoto, and et al. 2019. "Long-Term Observation of Atmospheric Speciated Mercury during 2007–2018 at Cape Hedo, Okinawa, Japan" Atmosphere 10, no. 7: 362. https://doi.org/10.3390/atmos10070362
APA StyleMarumoto, K., Suzuki, N., Shibata, Y., Takeuchi, A., Takami, A., Fukuzaki, N., Kawamoto, K., Mizohata, A., Kato, S., Yamamoto, T., Chen, J., Hattori, T., Nagasaka, H., & Saito, M. (2019). Long-Term Observation of Atmospheric Speciated Mercury during 2007–2018 at Cape Hedo, Okinawa, Japan. Atmosphere, 10(7), 362. https://doi.org/10.3390/atmos10070362