The Belgian CTM had a comprehensive stratospheric chemistry and had been delivering operational 4D-Var chemical analyses and forecasts for several years prior to the start of this study. The Belgian operational chemical analysis and forecast system is known as BASCOE and the resulting coupled model was named GEM-BACH (GEM with the Belgian Atmospheric CHemistry). The reader should note, however, that in the years our study took place, only a tropospheric version of GEM (with top at 10 hPa) was operational, so we conducted, ourselves, the tropospheric-stratospheric meteorological assimilation using AMSU-A channels 11–14 for the stratosphere.
3.1. Meteorological Model
GEM is a two time-level semi-Lagrangian fully implicit non-hydrostatic grid point model [
24] which uses either uniform or variable horizontal-resolution grids [
59]. An Arakawa C discretization is used in the horizontal and a hybrid vertical coordinate with non-staggered finite differences is used in the vertical. The model solves for horizontal and vertical momentum, thermodynamics, continuity, and the advection of an arbitrary number of tracers. For the stratosphere, additional physical parameterizations were added including a non-orographic gravity wave drag (GWD) [
39,
40] scheme for representing the breaking of unresolved non-stationary waves which has a major impact on the middle-atmosphere general circulation. For the computation of radiative transfer the model uses a radiation scheme based on the k-correlated distribution method [
60] which is an efficient approach to compute the radiation using a limited number of absorption coefficients. The radiation scheme also uses an ozone climatology based on Fortuin and Kelder (1998) [
61] which is merged with HALOE observations above 0.5 hPa. Finally, a stratospheric water vapor parameterized is employed, via a simple methane oxidation mechanism [
62,
63]. The description and validation of the stratospheric extension of GEM is discussed in Charron et al. (2012) [
57].
GEM-BACH used here is configured to run in hydrostatic mode with a global uniform resolution of 1.5° × 1.5°, i.e., 240 × 120 grid points. The vertical domain extends to 0.1 hPa and uses 80 vertical levels, 27 of which are in the stratosphere. Such high vertical resolution is uncommon to most Global Chemistry Climate Models (GCCM’s) which is a distinctive feature of the coupled model GEM-BACH.
Figure 3 compares the vertical grid used in this study with the ECMWF vertical grid used back in 2005 (at the beginning of this study). Altitude and vertical grid spacing are estimated using log-pressure altitudes (
), where the surface pressure
is set to 1000 hPa and the scale height
H is set to 7 km.
A series of meteorology-only climate simulations at a resolution of 1.5°× 1.5° using the additional stratospheric parameterizations (outlined above) was performed and the results were compared against either ERA 40 climatology or observations (see
Figures S3–S5 in the Supplementary Material). We observed that the native run (a version of the model with a top at 0.1 hPa but with no stratospheric physical parameterizations), exhibit a tropical tropopause and polar winter stratosphere which were too cold while the summer pole stratosphere was too close to radiative equilibrium. But with these stratospheric parameterizations these issues were significantly corrected compared with the ERA 40 reanalysis. Corrections to the zonal winds were also observed with these additional parameterizations (
Figure S4 Supplementary Material), where the Hines GWD scheme reduced the mesospheric jets and the new radiation scheme intensified the zonal wind in the stratosphere. The representation of interannual variability in the zonal winds, obtained from a zonal wind time series in the tropics, was also reasonably well captured with these additional parameterizations (
Figure S5 in Supplementary Material).
3.2. Stratospheric Chemistry Model
The photochemical module that has been implemented in GEM-BACH is the one used by the Belgian Institute for Space Aeronomy (BIRA-IASB) which includes 57 species, interacting through 143 gas-phase reactions, 48 photolysis reactions and 9 heterogeneous reactions.
Table 1 gives the list of species and
Appendix A details the list of photochemical reactions.
Appendix B explains the physics of the photochemical reaction rates, the so-called
J values.
Appendix C gives the lower chemical boundary condition at 400 hPa, the level blow which the chemistry solver is not active. The chemical reaction rates and photodissociation rates follow the Jet Propulsion Laboratory compilation [
64]. A complete description of the stratospheric chemistry is not intended to be covered here, and we refer the interested reader to appropriate review papers (e.g., [
29]).
The rates for gas-phase and heterogeneous chemistry depend on temperature. A reaction between two molecules has a reaction rate
of the form
where
E represents the energy of activation,
R is the gas constant and
A the Arrhenius factor. Reactions involving three molecules can also be pressure-dependent and require more complex formulations. Reaction rates involving aerosols are generally expressed as
where the term in parentheses represents the molecular mean speed of the gas-phase molecules, which depends on temperature
T, the molecular mass
M, and the Boltzman constant
k.
is the aerosol surface area per unit volume and
is the reaction efficiency representing the probability that a reaction takes place following the collision of the molecule with the particle. Chemical rate coefficients are determined experimentally and tabulated for different conditions [
64].
Heterogeneous chemistry plays an important role, especially in polar regions, and has been explicitly taken into account in GEM-BACH (see
Table A2). Hydrolysis reactions on the surface of Stratospheric Sulfate Aerosols (SSA) contribute mainly to the removal of active nitrogen in the lower stratosphere. In polar regions, another important class of aerosols is Polar Stratospheric Clouds (PSC). Such clouds usually form from SSA particles and grow at cold temperatures from the uptake of water vapor and nitric acid (see [
65] for a review). In GEM-BACH, the surface area available for heterogeneous reactions is parameterized in a crude manner. Instead of using a costly detailed microphysical calculation, we used a climatology of SSA surface area densities (see
Figure S6 in Supplementary Material). Type II PSC particles (primarily composed of water ice) are set to appear at temperatures below 186°K with a surface area density equal to
cm
/cm
. Between 186°K and 194°K, they are replaced by Type Ia PSC particles (primarily composed of Nitric Acid Trihydrate, NAT) with the same surface area density. The parameterization of PSCs also incorporates the impact of PSC sedimentation on water vapor (dehydration) and gaseous HNO
3 (denitrification). Exponential loss is prescribed for these two species, with characteristic times of 9 days for water vapor (at the gridpoints where type II PSCs are present) and 100 days for nitric acid (at gridpoints where type Ia PSCs are present).
Since the onset of heterogeneous chemistry on PSC depends on temperature, it is important that the meteorological model can reach the threshold temperature required.
Figure 4 shows the 15-year average temperature over the South Pole region (defined as the area south of 60° S) as function of height and the day of the year. Of course some years are different from others, and the temperature is not completely uniform in the polar vortex, but
Figure 4 does indicate that the GEM model reaches temperatures below 190° C.
3.3. Chemical Solver
The photochemical module and solver used here follow closely those of [
66] used at the Belgian Institute for Space Aeronomy (BIRA-IASB). The chemical solver acts on number densities (expressed as molecules-
), not on mixing ratios as in transport. Ignoring the issue of advection for the moment, the chemical tendency on a model grid is of the form,
where
is the vector of number densities for
J chemical species,
is the production term and
is the loss term. Together they define,
, the chemical transformation operator for species
i. Equation (
6) is obtained by adding all the product and loss processes for each species (
) from the list of chemical reactions given in
Appendix A. This results in
J chemical tendency equations, fewer than the total number of chemical reactions
N,
. An example of such a procedure is given in Tables 1 and 2 of Yudin and Khattatov (2010) [
67]. The photochemical transformation Equation (
6) actually forms a set of stiff non-linear equations that span a wide range of chemical timescales. In principle this is best integrated with an implicit time-discretization scheme (e.g., Backward Euler scheme),
which ensures computational stability. However, to deal with the non-linearity of
, a linearization around the state at time
(up to second order) can be made,
where
is the Jacobian of the chemical production and loss terms, leading to a semi-implicit scheme of the form,
which, although it is not guaranteed to be stable, is usually stable in practice. The resulting equation is then linear. The actual numerical scheme that solves the chemistry is a Rosenbrock solver of third-order that is a variant and generalization of Equation (
9), where the time step is subdivided into several internal time steps
h (here 3) (see [
68,
69] Section 16.6, [
70]). This solver is made numerically stable through the specification of the coefficient of the Rosenbrock scheme [
71]. The Fortran code needed to apply the Rosenbrock solver for the chemical kinetic equations can be built by the Kinetic PreProcessor (KPP) [
72] that also determines the appropriate magnitude of
h based on a tolerance factor set as 0.1 in the current version of the model. The chemical solver is applied from the model lid to 400 hPa due to the lack of tropospheric chemistry in the model. In the three bottom layers, species mixing ratios are specified to a set of values taken from the SLIMCAT CTM [
4] and are shown in
Table A4 in
Appendix C. Species vertical fluxes are null at the model lid.
The execution of the chemistry solver with a semi-Lagrangian transport scheme proceeds as follows. First, a semi-Lagrangian advection is performed on all species by interpolating from the upstream (i.e., departure) points to compute the mixing ratio at the arrival point on the model grid. Please note that all species have the same upstream point and interpolation weights, which are calculated only once. This represents a significant computational savings for the chemical transport. The species mixing ratio is converted into number density and the photochemical tendency for each species is computed on each model grid point. Once the number densities are updated, they are transformed back into mixing ratios for another transport time step or for a call to the physics scheme. The transformation from number density to mixing ratio follows the expression , where T is the temperature, p the pressure, the universal gas constant, and the Avogadro number.
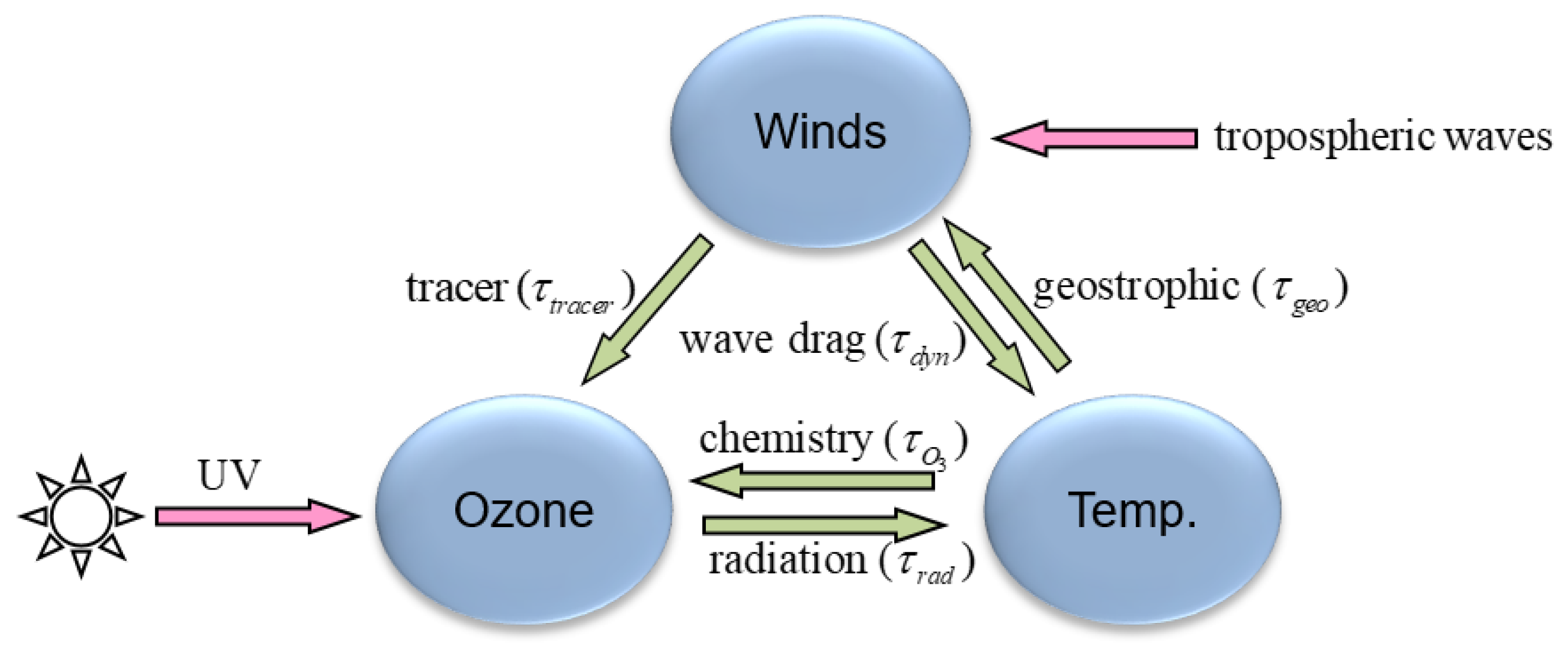
3.4. Model Coupling and Interface
Models are composed of several processes which are integrated either sequentially or in parallel (simultaneously). In sequential processing, for a given time step, the model state is updated after each process and provides input to the next process, until all processes are integrated. Sequential processing is also called time-splitting. In parallel processing, the tendencies of each process are computed simultaneously using the same initial model state. The updated state is computed from the sum of tendencies. Parallel processing is also called process splitting.
Parallel processing is appealing because of its simplicity and ease of implementation for coupled models. However, it has the disadvantage that the stationary solution of the time-discrete equation does not match the stationary solution of the time-continuous equation [
73,
74]. Sequential processing does not have this problem—it has the same stationary solution as the time-continuous equation. However, the transient solution depends on the order in which the different processes are integrated. The total error is minimized when the processes are ordered from the slowest process to the fastest [
75,
76].
The meteorological model GEM uses sequential processing and we have followed closely the same approach for the coupled meteorology-chemistry model configuration. In addition, we have adopted a modular design such that the chemistry component can be present (or not) through a chemical interface. This flexibility allows having a meteorology-only or meteorology-chemistry model configuration. However, this flexibility entails a small additional computational cost and maintenance, since some physics routines need to be duplicated and present in the chemical module. To make this clear let us begin by discussing what sequential processing would look like if chemistry were completely integrated with the physics module.
The processes in the meteorological model GEM are updated in the following sequence: 1—Radiation, 2—Advection, 3—Dynamics terms of meteorological variables using a semi-implicit scheme, 4—Surface fluxes and gravity wave drag (orographic and non-orographic), 5—Boundary layer processes and vertical diffusion, 6—Shallow convection, 7—Deep convection, and 8—Microphysics. A coupled meteorology-chemistry model has a 9th process—Chemistry, which involves very fast process that in principle should be solved implicitly but in practice we have chosen to use a semi-implicit approach using a Rosenbrock solver (see
Section 3.3). For the purpose of this discussion let us consider only those processes that involve both meteorological and chemical variables. For stratospheric chemistry those are: 1—Radiation, 2- Advection, 5—Vertical diffusion, and 9—Chemistry.
Let
represent the coupled (augmented) state vector, i.e.,
where
is the meteorological state vector and
the chemical state vector. Then the evolution that matters for the coupled state vector takes the form
where
represents the material derivative,
the radiation,
the vertical diffusion and
chemical processes.
The radiation
can be either offline or online with the prognostic chemical variables–in particular O
3. In the offline mode, greenhouse gases and a zonal-mean climatology of O
3 are given as input to the radiation. Our ozone climatology is based on Fortuin and Kelder [
61] and HALOE observations above 1 mb. Thus, the radiation process takes the form
Since radiation in the troposphere depends on cloud parameters that are diagnostic, and thus not advected, and also depends on temperature that is advected, it is desirable to compute radiation before advection in order to avoid any mismatch in the fields required as input. The radiation update thus operates on the initial state
to create an intermediate state
of the form
In a fully coupled ozone–radiation configuration that we will consider in Part II of this study [
1], the radiation process then takes the form
.
After radiation, advection is processed on both meteorological and chemical variables using
as the initial state. Without loss of generality, in a semi-Lagrangian scheme we can write the advection update as,
where
is the spatial coordinate and
the upstream displacement along the trajectory. The next process to consider is the vertical diffusion
that is applied on both meteorological and chemical state variables. The resulting update has the form
using diffusion coefficients
K computed from meteorological fields that are common to both meteorological and chemical variables. Finally, the coupled state is updated for the chemical processes,
with a resulting state
. Here in Equation (
16), the dependence on
is actually
, temperature and pressure. We also use the tilde
to emphasize that the Jacobian and chemistry production and loss terms are evaluated in terms of mixing ratio and not in terms of the number density as in Equation (
9).
Figure 5 displays the sequence of updates of the coupled (augmented) model state and which information is passed from meteorological to chemical modules (up and down arrows). The equal sign indicates that there are no changes and the horizontal arrows indicate changes due to a specific process update. To simplify, let us first discuss the case where there is no ozone–radiation coupling, i.e., let us ignore for now the dotted upward arrow.
First, infrared radiation does not change the chemical concentrations but does change the temperature. Then, the advection of chemical (prognostic) variables requires information about the displacement of the upstream point, , and the interpolation weights, , that are computed from the wind and the position of the upstream point. Next, applying the vertical diffusion on chemical variables requires sharing the diffusion coefficients, K, that are computed from the meteorology. Finally, chemistry requires information about temperature and pressure, but does not change the meteorological variables. When this last update is completed, the state at time is produced.
In a modular implementation, all processes that involve changes in chemical concentrations (in practice, with the exception of advection) can be included in a chemical module. The computation of the changes in chemical concentrations requires exchange of information through a chemical interface. For example, if we duplicate and include the vertical diffusion routine in the chemical module and pass the K coefficients to the chemistry module, the computation of the vertical diffusion of the chemical variables can be done in the chemistry module. Likewise, in principle, the advection of the chemical variables could be performed in the chemistry module if we duplicate the appropriate routines, but in practice it is easier (in terms of code maintenance), to simply pass the chemical concentrations to the advection routine that computes advection to all meteorological and chemical variables at once.
So far, the exchange of information is performed only one way, from meteorology to chemistry. It is then possible and easy to have a meteorology-only configuration separate from a meteorology-chemistry configuration, by simply allowing advection to be performed on an arbitrary number of variables.
The modularity of this approach can be preserved with sequential processing in the case of ozone–radiation interaction. Indeed, after the whole sequence of processes from advection to increasingly faster processes is completed, the prognostic ozone can be passed as the initial condition to the radiation scheme for the next model time step integration (see dotted upward arrow in
Figure 5).
Finally, in terms of computational resources, GEM-BACH is about five times slower than GEM (with no chemistry) on a uniform resolution 1.5° × 1.5°, i.e., 240 × 120 grid points, with 80 vertical levels, and running on 16 CPU (MPI 4 nodes, OpenMP 4 CPU). GEM-BACH transport (advection of 57 species) accounts for about 1/4 of the CPU time, the computation of the J-values for about 1/4 of the CPU time, and the Rosenbrock chemical solver about 1/2 of the CPU time.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
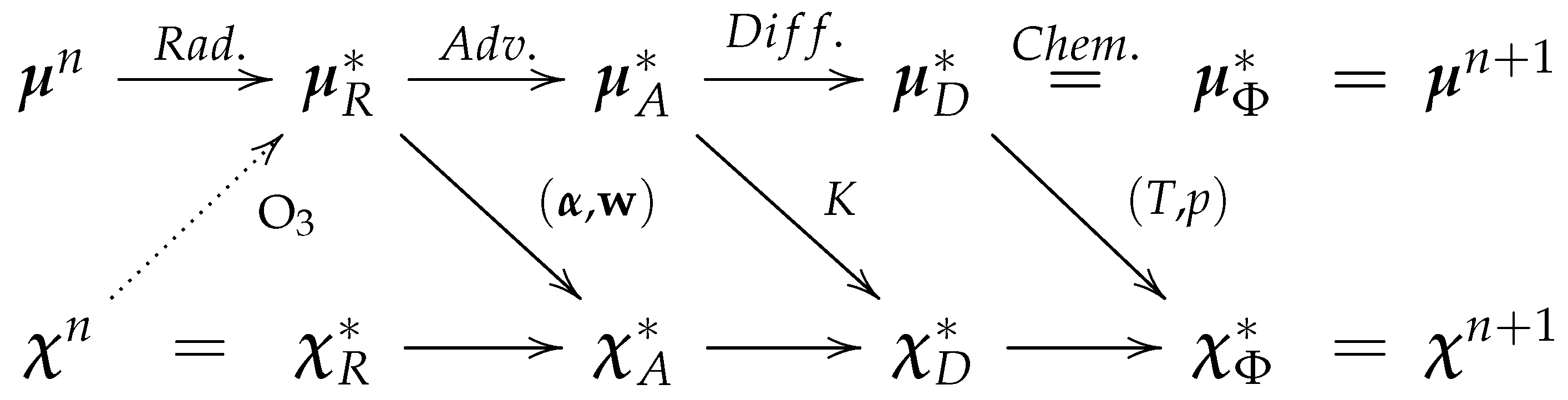{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













