
Chemical Composition of Gas and Particle Phase Products of Toluene Photooxidation Reaction under High OH Exposure Condition


 ,
,  , and
, and 
Abstract
:1. Introduction
2. Methodology
2.1. PAM OFR
2.2. Experimental Setup
2.3. Calibration of OH Concentration in PAM OFR
2.4. GC/EI-MS Analysis
3. Result and Discussion
3.1. Gas Phase Product Identification by PTR-MS
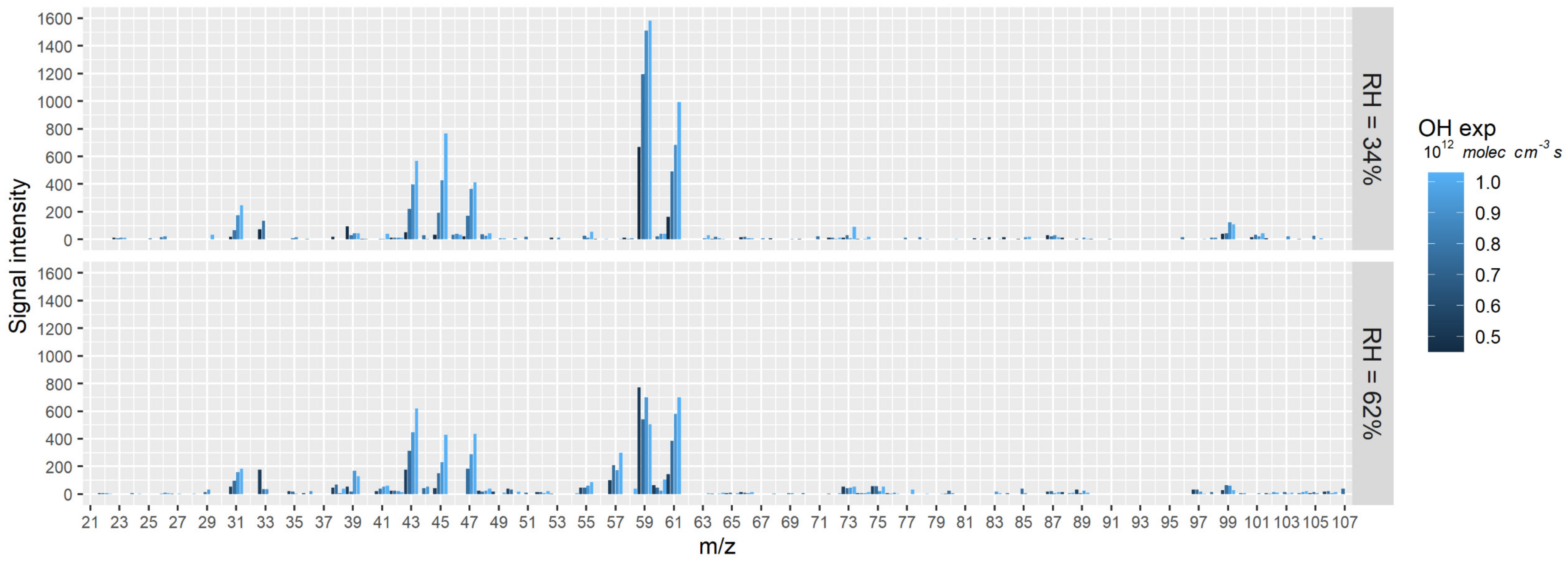
3.1.1. Peak Identification
3.1.2. Effect of OH Exposure on the Composition and Yield of Gas Phase Products
3.1.3. Effect of RH on the Chemical Composition of Gas Phase Products
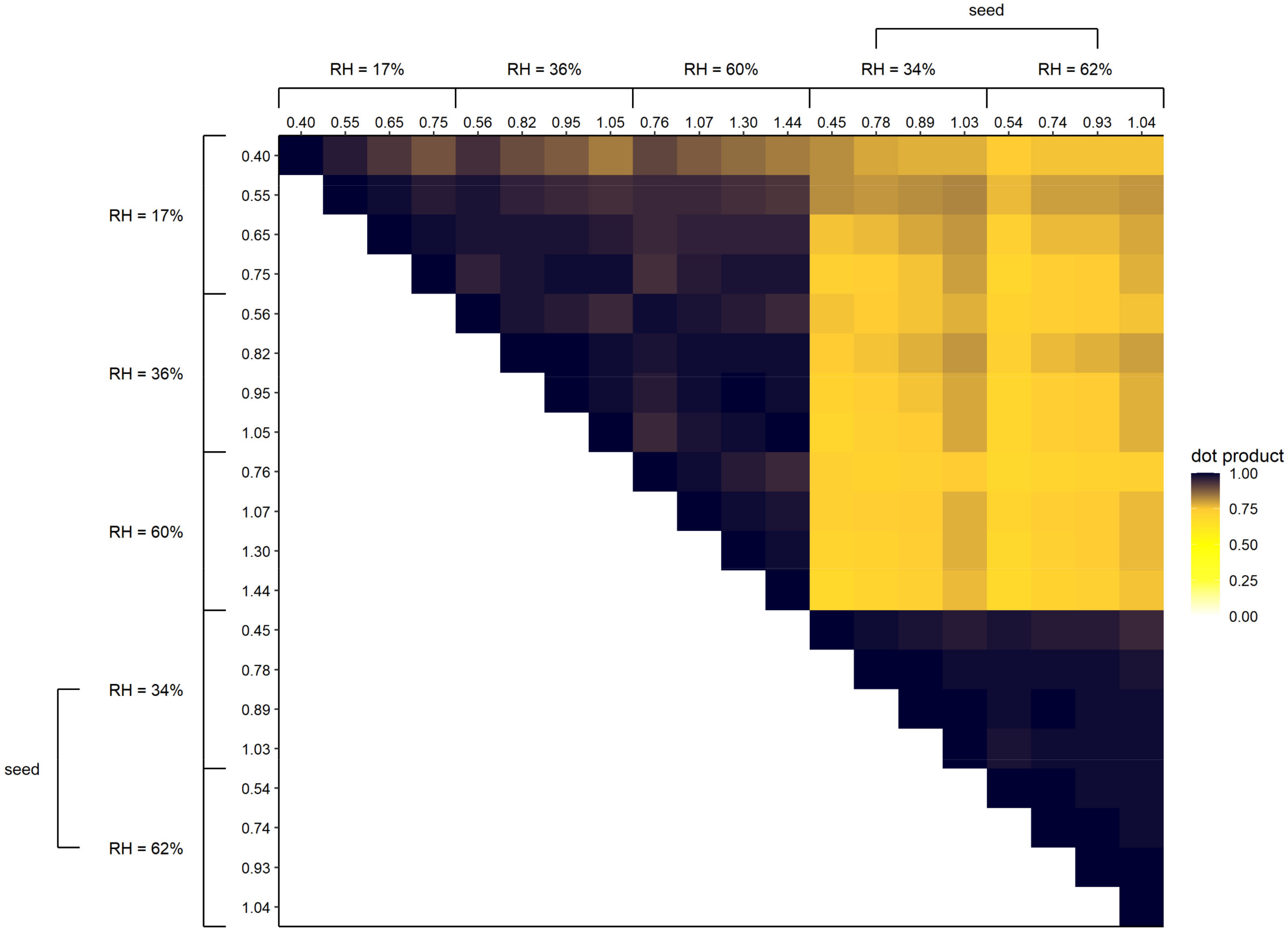
3.1.4. Effect of (NH4)2SO4 Seed
3.2. Particle Phase Products
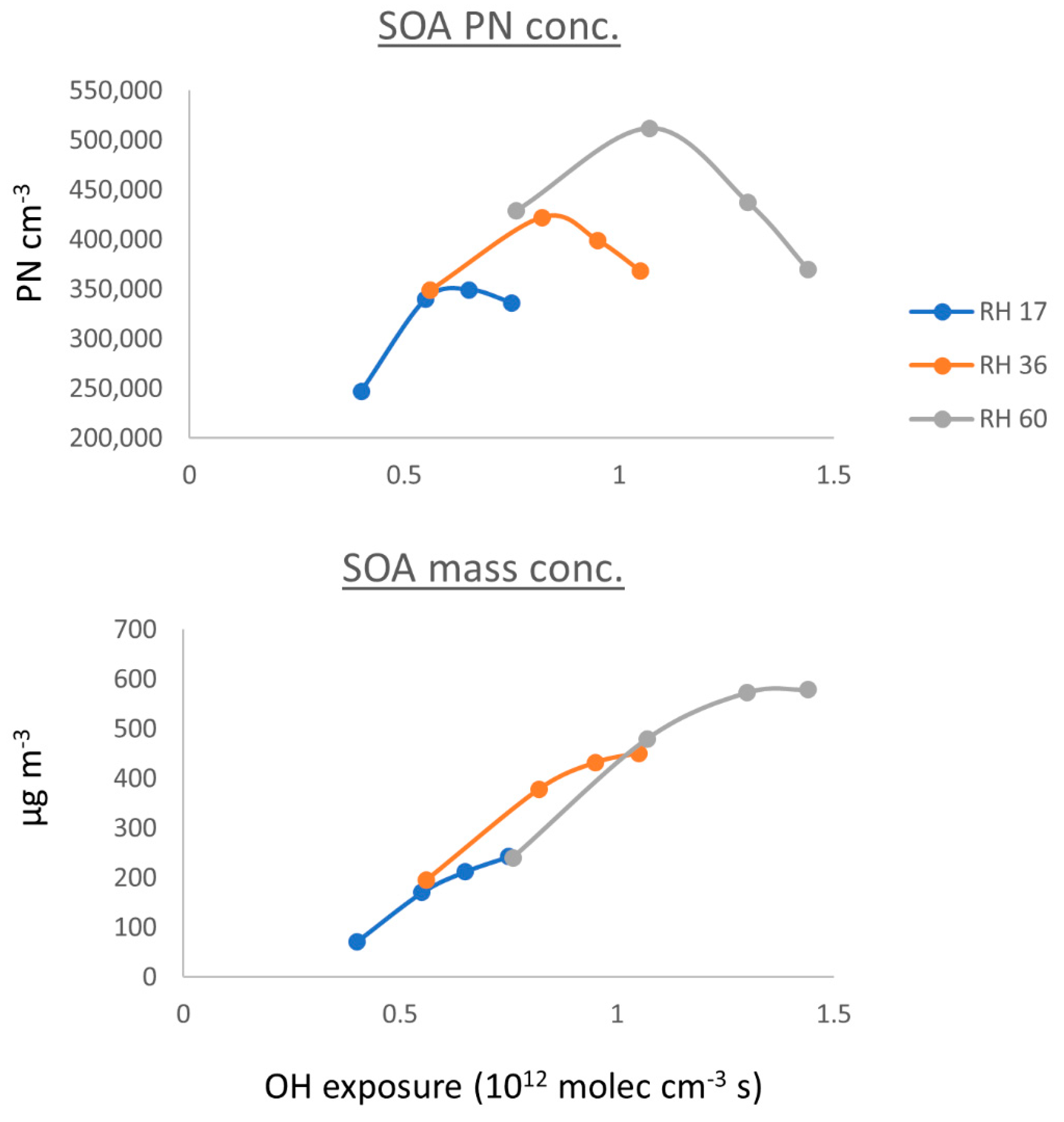
3.2.1. Number and Mass Concentration of SOA
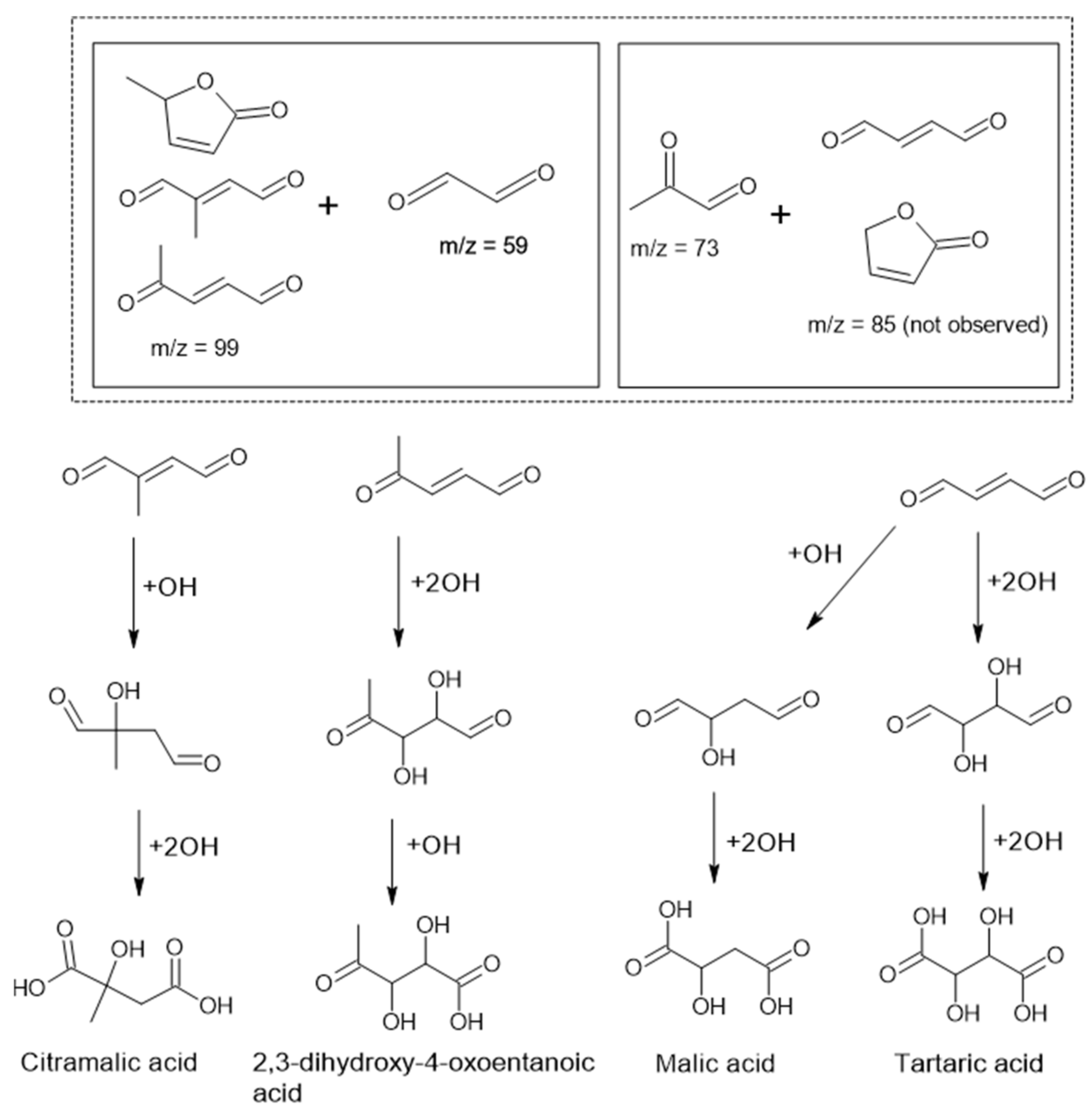
3.2.2. Chemical Characterization of Particle Phase Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, R.-J.; Zhang, Y.; Bozzetti, C.; Ho, K.-F.; Cao, J.-J.; Han, Y.; Daellenbach, K.R.; Slowik, J.G.; Platt, S.M.; Canonaco, F.; et al. High secondary aerosol contribution to particulate pollution during haze events in China. Nature 2014, 514, 218–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, M.; Cappa, C.D.; Fan, J.; Goldstein, A.H.; Guenther, A.B.; Jimenez, J.L.; Kuang, C.; Laskin, A.; Martin, S.T.; Ng, N.L.; et al. Recent advances in understanding secondary organic aerosol: Implications for global climate forcing. Rev. Geophys. 2017, 55, 509–559. [Google Scholar] [CrossRef] [Green Version]
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar] [CrossRef] [Green Version]
- Tuet, W.Y.; Chen, Y.; Fok, S.; Champion, J.A.; Ng, N.L. Inflammatory responses to secondary organic aerosols (SOA) generated from biogenic and anthropogenic precursors. Atmos. Chem. Phys. 2017, 17, 11418–11440. [Google Scholar] [CrossRef] [Green Version]
- Ng, N.L.; Canagaratna, M.R.; Zhang, Q.; Jimenez, J.L.; Tian, J.; Ulbrich, I.M.; Kroll, J.H.; Docherty, K.S.; Chhabra, P.S.; Bahreini, R.; et al. Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry. Atmos. Chem. Phys. 2010, 10, 4610–4641. [Google Scholar] [CrossRef] [Green Version]
- Davidson, C.I.; Phalen, R.F.; Solomon, P.A. Airborne Particulate Matter and Human Health: A Review. Aerosol Sci. Technol. 2005, 39, 737–749. [Google Scholar] [CrossRef]
- Valavanidis, A.; Fiotakis, K.; Vlachogianni, T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J. Environ. Sci. Health C 2008, 26, 339–362. [Google Scholar] [CrossRef]
- Ng, N.L.; Brown, S.S.; Archibald, A.T.; Atlas, E.; Cohen, R.C.; Crowley, J.N.; Day, D.A.; Donahue, N.M.; Fry, J.L.; Fuchs, H.; et al. Nitrate radicals and biogenic volatile organic compounds: Oxidation, mechanisms, and organic aerosol. Atmos. Chem. Phys. 2017, 17, 2103–2162. [Google Scholar] [CrossRef] [Green Version]
- Kroll, J.H.; Seinfeld, J.H. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 2008, 42, 3593–3624. [Google Scholar] [CrossRef]
- Ziemann, P.J.; Atkinson, A.R. Kinetics, products, and mechanisms of secondary organic aerosol formation. Chem. Soc. Rev. 2012, 41, 6582–6605. [Google Scholar] [CrossRef]
- Rollins, A.W.; Pusede, S.; Wooldridge, P.; Min, K.E.; Gentner, D.R.; Goldstein, A.H.; Liu, S.; Day, D.A.; Russell, L.M.; Rubitschun, C.L.; et al. Gas/particle partitioning of total alkyl nitrates observed with TD-LIF in Bakersfield. J. Geophys. Res. Atmos. 2013, 118, 6651–6662. [Google Scholar] [CrossRef]
- Deng, W.; Liu, T.; Zhang, Y.; Situ, S.; Hu, Q.; He, Q.; Zhang, Z.; Lü, S.; Bi, X.; Wang, X.; et al. Secondary organic aerosol formation from photo-oxidation of toluene with NOx and SO2: Chamber simulation with purified air versus urban ambient air as matrix. Atmos. Environ. 2017, 150, 67–76. [Google Scholar] [CrossRef]
- Robinson, A.L.; Donahue, N.M.; Shrivastava, M.K.; Weitkamp, E.A.; Sage, A.M.; Grieshop, A.P.; Lane, T.E.; Pierce, J.R.; Pandis, S.N. Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging. Science 2007, 315, 1259. [Google Scholar] [CrossRef]
- Liggio, J.; Li, S.-M.; Hayden, K.; Taha, Y.M.; Stroud, C.; Darlington, A.; Drollette, B.D.; Gordon, M.; Lee, P.; Liu, P.; et al. Oil sands operations as a large source of secondary organic aerosols. Nature 2016, 534, 91–94. [Google Scholar] [CrossRef]
- Lu, Q.; Murphy, B.N.; Qin, M.; Adams, P.J.; Zhao, Y.; Pye, H.O.T.; Efstathiou, C.; Allen, C.; Robinson, A.L. Simulation of organic aerosol formation during the CalNex study: Updated mobile emissions and secondary organic aerosol parameterization for intermediate-volatility organic compounds. Atmos. Chem. Phys. 2020, 20, 4313–4332. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Jimenez, J.L. Radical chemistry in oxidation flow reactors for atmospheric chemistry research. Chem. Soc. Rev. 2020, 49, 2570–2616. [Google Scholar] [CrossRef]
- Kang, E.; Root, M.J.; Toohey, D.W.; Brune, W.H. Introducing the concept of Potential Aerosol Mass (PAM). Atmos. Chem. Phys. 2007, 7, 5727–5744. [Google Scholar] [CrossRef] [Green Version]
- Tkacik, D.S.; Lambe, A.T.; Jathar, S.; Li, X.; Presto, A.A.; Zhao, Y.; Blake, D.; Meinardi, S.; Jayne, J.T.; Croteau, P.L.; et al. Secondary organic aerosol formation from in-use motor vehicle emissions using a potential aerosol mass reactor. Environ. Sci. Technol. 2014, 48, 11235–11242. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Lambe, A.T.; Saleh, R.; Saliba, G.; Robinson, A.L. Secondary Organic Aerosol Production from Gasoline Vehicle Exhaust: Effects of Engine Technology, Cold Start, and Emission Certification Standard. Environ. Sci. Technol. 2018, 52, 1253–1261. [Google Scholar] [CrossRef]
- Lambe, A.T.; Onasch, T.B.; Massoli, P.; Croasdale, D.R.; Wright, J.P.; Ahern, A.T.; Williams, L.R.; Worsnop, D.R.; Brune, W.H.; Davidovits, P. Laboratory studies of the chemical composition and cloud condensation nuclei (CCN) activity of secondary organic aerosol (SOA) and oxidized primary organic aerosol (OPOA). Atmos. Chem. Phys. 2011, 11, 8913–8928. [Google Scholar] [CrossRef] [Green Version]
- Molteni, U.; Bianchi, F.; Klein, F.; el Haddad, I.; Frege, C.; Rossi, M.J.; Dommen, J.; Baltensperger, U. Formation of highly oxygenated organic molecules from aromatic compounds. Atmos. Chem. Phys. 2018, 18, 1909–1921. [Google Scholar] [CrossRef] [Green Version]
- Palm, B.B.; Campuzano-Jost, P.; Day, D.A.; Ortega, A.M.; Fry, J.L.; Brown, S.S.; Zarzana, K.J.; Dube, W.; Wagner, N.L.; Draper, D.C.; et al. Secondary organic aerosol formation from in situ OH, O3, and NO3 oxidation of ambient forest air in an oxidation flow reactor. Atmos. Chem. Phys. 2017, 17, 5331–5354. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Chu, B.; Liu, J.; Liu, Y.; Zhang, H.; He, H. NOx promotion of SO2 conversion to sulfate: An important mechanism for the occurrence of heavy haze during winter in Beijing. Environ. Pollut. 2018, 233, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Lambe, A.T.; Chhabra, P.S.; Onasch, T.B.; Brune, W.H.; Hunter, J.F.; Kroll, J.H.; Cummings, M.J.; Brogan, J.F.; Parmar, Y.; Worsnop, D.R.; et al. Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield. Atmos. Chem. Phys. 2015, 15, 3063–3075. [Google Scholar] [CrossRef] [Green Version]
- Ortega, A.M.; Day, D.A.; Cubison, M.J.; Brune, W.H.; Bon, D.; de Gouw, J.A.; Jimenez, J.L. Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3. Atmos. Chem. Phys. 2013, 13, 11551–11571. [Google Scholar] [CrossRef] [Green Version]
- Lambe, A.T.; Onasch, T.B.; Croasdale, D.R.; Wright, J.P.; Martin, A.T.; Franklin, J.P.; Massoli, P.; Kroll, J.H.; Canagaratna, M.R.; Brune, W.H.; et al. Transitions from functionalization to fragmentation reactions of laboratory secondary organic aerosol (SOA) generated from the OH oxidation of alkane precursors. Environ. Sci. Technol. 2012, 46, 5430–5437. [Google Scholar] [CrossRef]
- Wang, Y.; Mehra, A.; Krechmer, J.E.; Yang, G.; Hu, X.; Lu, Y.; Lambe, A.; Canagaratna, M.; Chen, J.; Worsnop, D.; et al. Oxygenated products formed from OH-initiated reactions of trimethylbenzene: Autoxidation and accretion. Atmos. Chem. Phys. 2020, 20, 9563–9579. [Google Scholar] [CrossRef]
- Lambe, A.T.; Ahern, A.T.; Williams, L.R.; Slowik, J.G.; Wong, J.P.S.; Abbatt, J.P.D.; Brune, W.H.; Ng, N.L.; Wright, J.P.; Croasdale, D.R.; et al. Characterization of aerosol photooxidation flow reactors: Heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements. Atmos. Meas. Tech. 2011, 4, 445–461. [Google Scholar] [CrossRef] [Green Version]
- Rattigan, O.V.; Boniface, J.; Swartz, E.; Davidovits, P.; Jayne, J.T.; Kolb, C.E.; Worsnop, D.R. Uptake of gas-phase SO2 in aqueous sulfuric acid: Oxidation by H2O2, O3, and HONO. J. Geophys. Res. Atoms. 2000, 105, 29065–29078. [Google Scholar] [CrossRef]
- Mao, J.; Ren, X.; Brune, W.H.; Olson, J.R.; Crawford, J.H.; Fried, A.; Huey, L.G.; Cohen, R.C.; Heikes, B.; Singh, H.B.; et al. Airborne measurement of OH reactivity during INTEX-B. Atmos. Chem. Phys. 2009, 9, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.; Jenkin, M.E.; Wirtz, K.; Martin-Reviejo, M. Simulating the Formation of Secondary Organic Aerosol from the Photooxidation of Toluene. Environ. Chem. 2004, 1, 150–165. [Google Scholar] [CrossRef]
- Maleknia, S.D.; Bell, T.L.; Adams, M.A. PTR-MS analysis of reference and plant-emitted volatile organic compounds. Int. J. Mass. Spectrom. 2007, 262, 203–210. [Google Scholar] [CrossRef]
- Calvert, J.G.; Atkinson, R.; Becker, K.H.; Kamens, R.M.; Seinfeld, J.H.; Wallington, T.H.; Yarwood, G. The Mechanisms of Atmospheric Oxidation of the Aromatic Hydrocarbons; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Ji, Y.; Zhao, J.; Terazono, H.; Misawa, K.; Levitt, N.P.; Li, Y.; Lin, Y.; Peng, J.; Wang, Y.; Duan, L.; et al. Reassessing the atmospheric oxidation mechanism of toluene. Proc. Natl. Acad. Sci. USA 2017, 114, 8169–8174. [Google Scholar] [CrossRef] [Green Version]
- Arey, J.; Obermeyer, G.; Aschmann, S.M.; Chattopadhyay, S.; Cusick, R.D.; Atkinson, R. Dicarbonyl Products of the OH Radical-Initiated Reaction of a Series of Aromatic Hydrocarbons. Environ. Sci. Technol. 2009, 43, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Marcolli, C.; Canagaratna, M.R.; Worsnop, D.R.; Bahreini, R.; de Gouw, J.A.; Warneke, C.; Goldan, P.D.; Kuster, W.C.; Williams, E.J.; Lerner, B.M.; et al. Cluster Analysis of the Organic Peaks in Bulk Mass Spectra Obtained During the 2002 New England Air Quality Study with an Aerodyne Aerosol Mass Spectrometer. Atmos. Chem. Phys. 2006, 6, 5649–5666. [Google Scholar] [CrossRef] [Green Version]
- TSI. Aerosol Instrument Manager Sofware for Scanning Mobility Particle Sizer (SMPS) Spectrometer, User’s Manual; TSI: Shoreview, MN, USA, 2010. [Google Scholar]
- Loeffler, K.W.; Koehler, C.A.; Paul, N.M.; de Haan, D.O. Oligomer Formation in Evaporating Aqueous Glyoxal and Methyl Glyoxal Solutions. Environ. Sci. Technol. 2006, 40, 6318–6323. [Google Scholar] [CrossRef]
- Kleindienst, T.E.; Conver, T.S.; McIver, C.D.; Edney, E.O. Determination of secondary organic aerosol products from the photooxidation of toluene and their implications in ambient PM2.5. J. Atmos. Chem. 2004, 47, 79–100. [Google Scholar] [CrossRef]
- Lewandowski, M.; Jaoui, M.; Kleindienst, T.E.; Offenberg, J.H.; Edney, E.O. Composition of PM2.5 during the summer of 2003 in Research Triangle Park, North Carolina. Atmos. Environ. 2007, 41, 4073–4083. [Google Scholar] [CrossRef]
- Hu, D.; Bian, Q.; Li, T.W.Y.; Lau, A.K.H.; Yu, J.Z. Contributions of isoprene, monoterpenes, β-caryophyllene, and toluene to secondary organic aerosols in Hong Kong during the summer of 2006. J. Geophys. Res. Atmos. 2008, 113. [Google Scholar] [CrossRef]
- Sato, K.; Hatakeyama, S.; Imamura, T. Secondary organic aerosol formation during the photooxidation of toluene: NOx dependence of chemical composition. J. Phys. Chem. A 2007, 111, 9796–9808. [Google Scholar] [CrossRef]
- White, S.J.; Jamie, I.M.; Angove, D.E. Chemical characterisation of semi-volatile and aerosol compounds from the photooxidation of toluene and NOx. Atmos. Environ. 2014, 83, 237–244. [Google Scholar] [CrossRef]
- Ding, X.; Wang, X.-M.; Zheng, M. The influence of temperature and aerosol acidity on biogenic secondary organic aerosol tracers: Observations at a rural site in the central Pearl River Delta region, South China. Atmos. Environ. 2011, 45, 1303–1311. [Google Scholar] [CrossRef]
- Claeys, M.; Graham, B.; Vas, G.; Wang, W.; Vermeylen, R.; Pashynska, V.; Cafmeyer, J.; Guyon, P.; Andreae, M.O.; Artaxo, P.; et al. Formation of Secondary Organic Aerosols Through Photooxidation of Isoprene. Science 2004, 303, 1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.F.; Lewis, A.C.; Reynolds, J.C.; Carpenter, L.J.; Lubben, A. Investigating the composition of organic aerosol resulting from cyclohexene ozonolysis: Low molecular weight and heterogeneous reaction products. Atmos. Chem. Phys. 2006, 6, 4973–4984. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Tsona, N.T.; Zhang, Q.; Jia, L.; Xu, Y.; Du, L. Influence of relative humidity on cyclohexene SOA formation from OH photooxidation. Chemosphere 2019, 231, 478–486. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Smog Chamber | OFR | |
|---|---|---|
| Experimental timescale | Hours to day | Minutes to hours |
| Wall lose | Significant | Minimal |
| OH exposure | Hours to days | Days to weeks |
| Field work | Possible but not easy | Possible |
| Atmospheric relevance of oxidant and precursor concentrations | Yes | Possible for precursor, impossible for oxidant |
| Time for condensation and partitioning of products | Enough | Moderate |
| Chemical Structure | Molecular Weight | Average Mass on Each Filter (ng) | Previously Identified | |
|---|---|---|---|---|
| 2,3-Dihydroxy-4-oxopentanoic acid (DHOPA) |  | 148.1 g mol−1 | 35.4 | Kleindienst, Conver, McIver and Edney [39] |
| Citramalic acid |  | 148.1 g mol−1 | 269.7 | Lewandowski, Jaoui, Kleindienst, Offenberg and Edney [40], Hu, Bian, Li, Lau and Yu [41] |
| Malic acid |  | 134.1 g mol−1 | 68.5 | Sato, Hatakeyama and Imamura [42] |
| Tartaric acid |  | 150.1 g mol−1 | 78.6 | White, Jamie and Angove [43] |
| 2,3-dihydroxyglutaric acid |  | 164.1 g mol−1 | 56.2 | New product |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, Y.-S.; Chan, M.-N.; Poon, H.-Y.; Tan, Y.; Lee, S.-C.; Li, J.; Ho, K.-F. Chemical Composition of Gas and Particle Phase Products of Toluene Photooxidation Reaction under High OH Exposure Condition. Atmosphere 2021, 12, 915. https://doi.org/10.3390/atmos12070915
Lau Y-S, Chan M-N, Poon H-Y, Tan Y, Lee S-C, Li J, Ho K-F. Chemical Composition of Gas and Particle Phase Products of Toluene Photooxidation Reaction under High OH Exposure Condition. Atmosphere. 2021; 12(7):915. https://doi.org/10.3390/atmos12070915
Chicago/Turabian StyleLau, Yik-Sze, Man-Nin Chan, Hon-Yin Poon, Yan Tan, Shun-Cheng Lee, Jianjun Li, and Kin-Fai Ho. 2021. "Chemical Composition of Gas and Particle Phase Products of Toluene Photooxidation Reaction under High OH Exposure Condition" Atmosphere 12, no. 7: 915. https://doi.org/10.3390/atmos12070915
APA StyleLau, Y. -S., Chan, M. -N., Poon, H. -Y., Tan, Y., Lee, S. -C., Li, J., & Ho, K. -F. (2021). Chemical Composition of Gas and Particle Phase Products of Toluene Photooxidation Reaction under High OH Exposure Condition. Atmosphere, 12(7), 915. https://doi.org/10.3390/atmos12070915